
First Workshop on Nanomedicine for Infectious Diseases of Poverty, 27-31 March 2011, Magaliesberg, South Africa
10 May 2011. Related: Conference reports, Conference index, Nanomedicine 2011.
Simon Collins, HIV i-Base
Introduction
An international workshop on nanomedicine for infectious diseases related to poverty was held in Magaliesberg, South Africa from 27-31 March 2011.
The meeting was organised by Dr Hulda Swai, chair of the nanotechnology programme at the Council for Scientific and Industrial Research (CISR), a multidisciplinary science and research institute established in 1945 and funded by the South Africa Department of Science and Technology and Economic Commission for Africa (ECA). CSIR is one of two SA government-funded centres with nanomedicine programmes (the other is MINTEC). About 70 delegates from 20 countries attended the workshop.
Nanotechnology is being used in many different aspects of medical care including therapeutics, new drug delivery systems, diagnostics, imaging and surgical procedures. This article principally focuses on new formulations of drugs and drug delivery systems. Nanomedicine – the application of nanotechnology to medicine – has a relevance for HIV and related infections that rarely gets much attention.
Most of the focus on pipeline drugs for HIV and TB is on new compounds or new oral formulations of already approved drugs. However, for the last fifteen years various laboratories have been working with nanoformulations of antriretrovirals, though none have yet resulted in new medicines.
Size and scale
The simplest explanation of nanomedicine is based on a size ranging from 10-100 nm, though the EU definition has an upper range of 1000 nm. One nanometer is one-billionth of a meter (the width of about five atoms). See Table 1.
At this scale particles have different physical properties relating to surface to volume ratio, surface tension, surface charge and quantum dot effect and this can enhance drug bioavailability and solubility. Engineering molecules to target specific cellular and tissue targets has the potential to overcome barriers to sanctuary sites including the blood-brain barrier. Although nanotechnology is generally associated with the concept of the smaller particles, nanomedicine is actually based on drug formulations that are larger that pure drug molecules.
| Factor of 10 | Metric size | Example size | Comment |
|---|---|---|---|
| 10 (0) | 1 m (1000 mm) | a child | |
| 10 (-1) | 100 mm | an orange | |
| 10 (-2) | 10 mm | a marble | |
| 10 (-3) | 1 mm (1000 μm) | a pin head (1mm) grain of salt and an amobea (both ~500 μm) |
|
| 10 (-4) | 100 μm | human egg (130 μm) hair width (100 μm) |
|
| 10 (-5) | 10 μm | Red blood cell (8 μm) chromosome (7 μm) bakers yeast (3 x 4 μm) mitochondrion (4 x 0.8 μm) E. coli bacterium (3 x 0.6 μm) |
cells |
| 10 (-6) | 1 μm (1000 nm) | measles virus (220 nm) HIV (120 nm) influenza (130 μm) phage (bacteria virus) (70 x 200 μm) |
viruses,bacteria |
| 10 (-7) | 100 nm | hepatitis virus (45 nm) rhinovirus (30 nm) ribosome (30 nm) |
viruses,bacteria |
| 10 (-8) | 10 nm | antibody (12 nm) tRNA (7 nm) haemoglobin (6.5 nm) |
molecules |
| 10 (-9) | 1 nm (1000 pm) | adenine (1300 x 760 pm) methionine (1100 x 700 pm) glucose (900 pm) carbon atom (340 pm) water molecule (275 pm) |
molecules |
| 10 (-10) | 100 pm | hydrogen atom (100 pm) | atoms |
| 10 (-15) | nucleus |
Note: relative size from 10 (0) to 10 (-10) is similar to comparing the size of the world to a golf-ball.
Attaching compounds to larger molecules or encapsulating them inside other molecules can deliver a drug to the target site more accurately. This can overcome one of the main limitations of current oral formulations, where over 90% of medicines are excreted unused.
These improvements include:
- Better bioavailability, as an example this could be achieved by designing formulations that overcome hydrophobic or hydrophilic properties of individual molecules.
- Reducing drug wastage by overcoming protein binding and during oral absorption, where >90% of the active compound of antiretroviral drugs are cleared by blood filtration through the liver or kidneys before it is able to act on HIV.
- More targeted delivery should reduce the quantity of raw materials needed. This, is turn, has the potential to have the biggest impact on drugs used in resource-limited settings. Even though the drugs are much cheaper in poorer countries a much higher percentage of the costs is related to the active pharmaceutical ingredients (API).
- Reducing toxicities related to the metabolism of current oral formulations. For example, if a nanoformulation is designed to increase active drug levels inside cells while keeping blood levels low this has the potential to reduce toxicities related to systemic drug levels.
- Sanctuary site penetration by developing formulations that target immune cells that can cross the blood-brain barrier. In a similar way molecules may be designed to use cells to evade drug transporters such as P-gp that limit penetration of other sites.
However, while the potential benefits are promising, they also bring significant challenges to safety and regulatory approval.
Review of nanomedicine
The conference opened with a plenary lecture from Professor Ruth Duncan from Cardiff University, who has been a leading researcher for over 35 years in community-based research (Cancer UK), industry and academia. This talk emphasised the diversity of expertise needed from different fields including pharmacology, chemistry, medicine, ethics, health policy and politics.
Material science has been reducing the size of everything. Nanotechnology has been a rapidly evolving field that includes a top-down approach (where compounds are reduced) and a bottom-up approach (assembling polymer materials from the bottom up). Advanced drug delivery systems have been the focus of research for over 40 years. Nanomedicines can prolong action using new control/release technology, can target specific organs, cells, or organ space in an organelle, and improve bioavailability, including penetration of the blood-brain barrier.
Systemic distribution of most drugs involves a high level of dilution throughout the body and low concentrations at the active target. Nano-sized particles have different pharmacokinetics, delivering drugs by endocytosis and phagocytosis (cell engulfment) rather than passing directly through a cell wall. In addition to advancing drug delivery some nanoparticles are active in their own right. There are multiple targets through the lifecycle of infection and progression, including latent infection, each using different strategies.
The definition of nanomedicine incorporates engineering tools used outside the patient, biomedical and medical materials, imaging, and drug delivery and formulation. Designing nanomedicine should be easy. Starting with a target disease and product profile it should be possible to choose technologies and benchmark these against current treatment, with a clear stop-go development. Good lead candidates then require five years preclinical trials before getting to human studies. These drugs need a constant awareness of good laboratory practice (GLP), good manufacturing practice (GMP), good clinical practice (GCP) and ethics.
While nanomedicine has an unprecedented opportunity for invention, the lack of current knowledge across the field risks newer researchers repeating mistakes that earlier research has overcome. Against this is a caution that over exaggeration of the benefits will raise unrealistic expectations. Professor Gordon highlighted a limitation from the commercial focus on the target, using genomics and low molecular weight compounds, where 95% of compounds fail, probably because pharmacokinetics is not sufficiently important earlier in the development phase.
The first nanomedicines developed in the1990s based on liposome formulations are at the end of their patent life and are now coming through as generics. However each nanomedicine construct has a different design and route of delivery and every drug must be reviewed separately due to the specific challenges of manufacturing and constructing lipid-based nanoformulations. This field is more complex than generic antiretrovirals for example, where reproducing a compound that has similar pharmocokinetic properties is closely associated to similar safety and activity. From a safety perspective, producing new molecules, each piece (polymer, linker, etc) needs to be designed, requiring preclinical research before human studies. Many polymers for example are rejected for safety. Non-biodegradable polymers can accumulate, especially with long exposure (potentially lifelong) treatment. Pegylation itself covers a wide range of polymers. Currently the EMA opinion on generic formulations of Doxil etc is that bioequivilence does not exist for a liposome, only bio-similar properties and that pegylation on surface of liposome cannot just be duplicated as generic.
This complexity involves molecules travelling in the bloodstream at hundreds of miles an hour and the challenge is how to stick your arm out to catch them at the right site. Shape and density are essential aspects of design including ionisation of polyelectrolytes and changes in cellular behaviours. Linkers need to be stable and release drug at the right time. Activating compounds can behave differently, including by sex. All of parts of the system will go somewhere and these need to be accounted for to ensure patient safety.
The importance of products used in clinical trials and subsequently approved to have passed good GLP and GMP was introduced as a theme that would be frequently revisited throughout the workshop.
Historical perspective of nanomedicines in cancer treatment
Dr Theresa Allen from the University of Alberta, Canada, and Alberto Gabezon from the Shaare Zedek Medical Center, Jerusalem gave lectures on the historical perspective of nanotechnology drawing on the impact that it has had on cancer therapy.
Although molecules on a nanoscale have been known since 1900 the term is associated with a lecture given by the Nobel physicist Richard Feynman in 1959 setting two challenges based on the theory that atoms on a small scale behave like nothing on a large scale.
Nanotechnology based products already in common use include sunscreens (nanomolecules of titanium dioxide and zinc oxide are translucent, not white), self cleaning windows, pharmaceutical inks printing on medicines, and fabrics and coatings treated to variously repel dirt, water, bacteria, fungus or creasing.
In medicine, applications include imaging, diagnostics, drug delivery and therapeutic agents able to cross biological barriers. Nanomedicines deliver proteins, plasmid DNA, siRNA and antisense, enhanced permeability and retention (EPR) effect for solid tumours (where molecules target tissues with increased permeability and are only released in inflamed tissue and tumour damaged sites).
Nanomedicines can improve properties of existing drugs, reducing toxicity and improving bioavailability (poor bioavailability currently wastes an estimated 68 billion dollars annually).
Liposome and other lipid-based systems are the first and most common lipid medicines. They are simple, self-assembling, with proven safety, with flexibility in terms of particle size, release rates and bio distribution. A principal behind nanoformulations is that a drug changes its biological pharmacokinetics and distribution properties, generally being protected from degradation and metabolism, and only behaving as a regular molecule when released as free drug.
Examples of widely used nanomedicines include Doxil (a pegylated liposome encapsulation formulation of doxorubicin used to treat Kaposis sarcoma [KS] and ovarian cancer), the antifungal AmBisome (a liposomal formulation of amphotericin-B) and pegylated interferon (used with ribavirin to treat hepatitis C), Abraxone (a nanoformulation of paclitaxil used to treat metastatic breast cancer), Rapamune (formulation of sirolimus that is milled down and stabilised to become soluble) and Genoxol PM (a micelle formulation of paclitaxil). See Figure 2.
| Name | Year | Clinical indication | Comment |
|---|---|---|---|
| Doxil | 1995 | Kaposis Sarcoma (KS)Ovarian cancer | Pegylated liposome encapsulation formulation of doxorubicin |
| AmBisome | 1997 | Fungal infections, some antiprotazoal activity. | Liposomal formulation of amphotericin-B |
| PegIntron and Pegasys | 2001, 2002 | Hepatitis C | Pegylated interferons. |
| Abraxone | 2005 | Metastatic breast cancer | Nanoformulation of paclitaxil. |
| Genoxol PM | 1999 | As paclitaxil: ovarian, KS, breast cancers. | Micelle formulation of paclitaxil. |
| Rapamune | 2000 | Immunosuppressant to prevent organ graft rejection. | Formulation of sirolimus that is milled down and stabilised to become soluble. |
The first oncology nanomedicine was Doxil, approved in 1995 as a treatment for AIDS-related KS under fast track orphan drug designation with development time of less than ten years. Subsequent approvals for relapsed ovarian cancer, metastatic breast cancer and as a substitute for doxorubicin resulted in Doxil becoming a commercial blockbuster drug.
Doxil is a 100 nm diameter surface-graft pegylated long-circulating formulation where the active drug stays in a liposome, increases the concentration in sold tumours and slow releases in tumour cells. If ligands are added to particles, the resulting package can enter a cell in larger quantities and where it is then released.
Compared to the original doxorubicin, Doxil dramatically reduced clearance by 1000-fold (extending the half-life to 50-70 hours) and increased drug accumulation in tumour tissue (approximately 80 ug vs 0.5 ug in skin a few millimetres away). This enabled a 4-fold lower dose of Doxil to have a 30% higher cure rate and reduction tumour volume. Unfortunately, in this early example, side effects were not always reduced and a higher rate of palmar-plantar erythema rash occured in some patients.
Nanomedicine can also incorporate combination chemotherapy in single constructs using drugs that use different mechanisms and that have non-overlapping side effects. It is possible to mix two liposomes or to add multiple targets on each (for example to target to nothing, CD20, CD19 or both and produce additive effects). In oncology, combination approaches to target and kill blood vessels in tumour tissue starve the tumour of oxygen and nutrients (though peripheral tumour cells survive). New blood vessels in a tumour are damaged, disorganised and with incomplete endothelial lining, allowing particles up to 400 nm to penetrate. An early paper in 1982 showed the potential of liposomes to carry adriamycin in mice. Conventional liposomes reduced cardiovascular risk but did not target liver tumours and were leaking drug in blood. Changing the lipid concentrations in new formulations changed pharmacokinetics with a 20-fold increase in concentrations in the tumour and 4-fold reduction in spleen, highlighting the importance of liposome design.
Targeted siRNA or antisense molecules can be effective but the high charge restricts transfer across cell membranes. Adding ligands enables a Trojan horse effect so a drug can cross cell membranes. For example, antibody-targeted coated cationic liposomes can be used to inhibit anaplastic lymphoma kinase (ALK) in neuroblastima-bearing mice with anti-GD2-targetted CCLs entrapping ALK-siRNA. Free siRNA cannot cross cells but can in a nanoformulation.
A brief history (and reality check) of the time it has taken to develop nanomedicines starts with the first description of liposomes in the early 1960s, which became the foundation of many formulations. Liposomes are 100 nm nanoparticle spheres (visicles) consisting of a phospholipid shell where one end is hydrophobic (usually the outer surface) and one is hydrophilic, enclosing an internal water phase. Drug molecules can be either encapsulated in the water phase or entrapped in the lipid shell. See Figure 3.
Figure 3: Cross section of liposome used for drug delivery
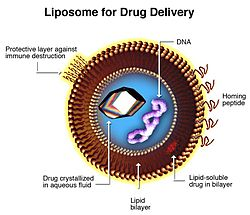
Source: http://en.wikipedia.org/wiki/Liposome
Further research led to the development of stealth liposomes in 1987 that used polyethylene glycol (PEG) studding on the outer coating to evade the mononuclear phagocyte immune system. AmBisome was approved in 1990, SMANCS (styrene maleic acid neocarzinostatin) in 1993 and Doxil in 1995. Nano-based drug delivery systems began entering mainstream medicine after 2005 and by 2011 there are over 38 nano products on market (worth $6.8 billion annually) with generic systems on the horizon.
The challenge for nanoformulations is to get an appropriate drug using appropriate delivery system that is the right size (not damaging other tissue) with low toxicit, for sufficient duration to solve an unmet medical need.
Crucially this needs to be at a sufficiently low cost to make it usable in a way that is safer and more effective than current treatments. Involvement of Indian companies has successfully radically reduced costs in many areas. In addition to reducing antiretroviral treatment from $10,000 to under $100 per year, the cost of hepatitis vaccinations have been reduced from $20 to $1, psoriasis treatment has been reduced from $20,000 to $100 and cataract surgery is carried out at 1% of the UK cost.
While the political aspects of new technology for infectious diseases have a high level of collaboration between countries at end stage distribution, the level is low for the early stages of research and development. The research presented at this workshop is going some way to address this.
A nanoformulation of paediatric efavirenz
Dr Alejandro Sosnik from the University of Buenos Aires discussed the potential applications of nanotechnology for paediatric formulations of HIV medicine. [4]
HIV in rich countries has become a largely manageable adult disease with more than 25 antiretroviral drugs and early diagnosis and access to HAART has reduced mother to child transmission to rates less than 1%. However, in most poorer countries HIV remains an acute disease with an estimated 1000 new paediatric infections globally each day, mostly in sub-Saharan Africa. Only 10% of children with HIV currently have access to treatment, and this drops to 2% in some regions.
Paediatric treatment in all countries is limited, with only 12 approved paediatric formulations. In particular, the lack of appropriate formulations, difficulties of dose adjustment, pill swallowing, and poor taste add to the challenges of enabling children to achieve the similar benefits from the advances seen in adult HIV care.
Dr Sosnik presented an example of a potential nanoformulation for efavirenz from his research group. Although a paediatric liquid formulation of efavirenz is produced by BMS this is not available globally and the indication is for children older than 3 years (and greater than 13 kg). The current oral formulation has lower bioavailability (by 40-45%) than capsule formulations and tastes like liquid Vaseline confirmed by associated weight-loss and diarrhoea. Interpatient and intrapatient bioavailability varies by 55-58% and 19-24% respectively.
The research goal and important unmet medical need is to develop a highly concentrated aqueous formulation with higher bioavailability, working with existing polymers like PEO-PPO polymeric micelles (poloxamers andoloamines) that are already commercially available in a broad spectrum of molecular weights and that already have a proven safety record (ie ethylendiamine).
So far, efavirenz-loaded micelles using surface aptamers to target CD4 cells have been produced in an oral solution and animal bioavailability data has been published. [5 – 9]. A micelle is an aggregate of compounds that lower the surface tension of a liquid and that are evenly dispersed.
In male rats the efavirenz-loaded micellar formulation was compared to that of a suspension prepared with the content of efavirenz capsules in 1.5% carboxymethylcellulose PBS solution (pH 5.0), and an EFV solution in a medium-chain triglyceride (Miglyol 812).
This formulation showed that the encapsulation of efavirenz, which is otherwise poorly water soluble, into polymeric micelles of different poly(ethylene oxide)poly(propylene oxide) block copolymers significantly improves oral bioavailability and reduces the interindividual variability.
The solution has an improved taste, using only excipients approved by the FDA, and requiring less API has the potential to lower production costs.
A protocol has already been approved by an ethics committee in Buenos Aires for first in-human pharmacokinetic studies in HIV-negative volunteers, which if successful will progress to studies in HIV-positive adults and then HIV-positive children.
One of the discussion points after the presentation included the use of the formulation first in HIV-positive adults. This has been by the research group and not being linked to clinical HIV centres in Argentina. Expanding on this, Dr Soznik explained, funding is difficult, industry studies are dominant. We need to now see if human exposure is similar to rats. Further animal studies might help but these are too expensive.
Other antiretroviral nanoformulations
Two other research groups at the workshop also presented data on formulations of antiretrovirals that are already approved, though none have yet been used in human studies.
Dr Lebogang Katata from CSIR in South Africa presented a poster on spray-dried efavirenz nanoparticles that was previously shown at the IAS conference in Vienna last year. [10, 11]
In this process, efavirenz is encapsulated in a polycaprolactone (PCL) polymer by a double emulsion spray drying technique using two organic solvents. The nanoparticles have an average size of 220.6 ± 0.950 nm when using ethyl acetate and 372.1 ± 19.96 nm using dichloromethane. This formulation also overcomes the hydrophobic nature of efavirenz to improve bioavailability and it has met other manufacturing standards including encapsulation efficiency and a smooth particle surface. It also results in prolonged release compared to formulations of free drug (suggesting weekly dosing might be possible). The group have also produced formulations of other antiretrovirals including AZT and d4T and plan combination formulations. But the timeline for human studies that were due to start this year is likely to be several years away.
A poster from Dr Helanie van der Merwe and colleagues at the North-West University, Potchefstroom, South Africa presented results from a third technology for producing nanoparticles of antiretrovirals. [12]
This research group has encapsulated abacavir and lamivudine (3TC) in visicles using a technology called pheroid, that resulted in higher bioavailability in in vitro studies.
Professor Anne Grobler from North-West University explained pheroid technology in an oral presentation. [13]
This talk started with an example of South African drug development of Exorex. This is a coal tar preparation for psoriasis that was initially developed by an American individual for his personal treatment and was then developed by the University into a global treatment, with the advantage of greater efficacy from using a much lower percentage of coal tar (1% vs 5%). [14]
Pheroids use a form of colloidal transport (usually at 100 nm size but sometimes larger) using long chain fatty acids, modified in different ways, ie with pegylation to have with hydrophilic tails. The technology is apparently easy, and inexpensive but it was originally difficult to predict the type of pheroid because it was not designed working with the active compound. All fatty acids are currently being taken safely by humans and the technology allows packaging of more than one drug in each vesicle.
In addition to abacavir and lamivudine the group have produced Pheroid formulations of antimalarial [15] and antituberculousis drugs [16, 17] that in preclinical development consisted of comparative in vitro and animal efficacy, bioavailability and pharmacokinetic studies and limited toxicology studies.
These studies reported enhanced rates of in vitro efficacy and in vivo bioavailability of first and second-line antimalarial compounds (including chloroquine, mefloquine, artemether and artesunate) and extended in vivo pharmackinetics and efficacy studies with formulation of tuberculosis drugs in mice.
Formulations of anti-TB medications
The CSIR research programme for formulations of TB medications is more advanced than that for HIV. Dr Rose Hayeshi, expanded on this programme. [18]
The group have encapsulated four first line anti-TB drugs (isoniazid, rifampicin, ethambutol and pyrazinamide) with an encapsulation efficiency varying from 50-65% in particles of 250-400nm, using a multiple emulsion spray-drying technique.
The polymer used is poly(lactide-co- glycolide), (PLGA). The particles are taken up by macrophages in vitro indicating feasibility of intracellular drug delivery. Studies in mice using fluorescently-labelled PLGA nanoparticles indicated distribution to a broad diversity of tissues including macrophages of the peritoneum cell exudates that cross the blood brain barrier. Safety in mice after up to 10 days exposure was supported by histopathology on all major tissues including the spleen, lungs, kidney, liver, spleen, heart and the brain, which found no evidence of lesions.
Pharmocokinetic studies in mice showed that the drugs were released over a period of six days and the minimum inhibitory concentration for rifamicin and izoniazid was maintained over this period. Pharmacokinetic curves are similar to those of free drug, with an initial increase from drug on the outside of the molecule followed by slow release compared to free drug.
A study in six TB-infected mice dosed daily for four weeks showed a reduction in colony forming units in spleen, liver although there is a need to target actively to lungs. When repeated over nine weeks an improved pathology was observed in the lungs. The nanoformulation is a weekly dose compared to current requirement for strict daily dosing.
The research group are now looking at scale-up plans for all formulations, improved preclinical, dose formulation and design and then first clinical studies. Currently single formulations are used but combinations are planned and the group is working with compounds held by the TB Alliance.
The discussion after this presentation focused on the need to clarify the mechanism and route of administration for TB drugs. This included whether the controlled release is in blood, tissues, or the cells. Oral formulations are difficult to target to cells, and compounds often need IV administration, which is not practical in resource-limited settings.
Aptamers in nanomedicine
Dr Makobetsa Khati, also based at the CSIR explained the utility of nanoaptamer biconjugates against infectious diseases with a focus on HIV. [19]
DNA or RNA aptamers were first developed in 1990 and are usually short strands of artifical oligonucleotides that are selected in vitro using SELEX (systematic evolution of ligands by experimental enrichment).
At a size that is less than 10 nm radius they can increase tissue specificity with minimal impact on the size of the formulation. Current uses include delivery of drugs to treat age related macula degeneration. Aptamers have the molecular recognition properties of monoclonal antibodies in terms of their high affinity and specificity. They are chemically simple, easy to make, have high target specificity, are non-toxic and non-immunogenic. Aptamers are used in many of the formulations already discussed in order to identify target cells for nanoformulations.
As aptamers can block entry this group has worked with oligonucleotides that are active against gp120. [15] The aptamer doesnt affect cell metabolism with toxicity, immune of cardiovascular cells. Over 80% of cells were sensitive with IC50 <1 nm and non-toxic at concentrations over 1000 nm.
In humanised mice Neff and colleagues showed reduction in viral load and a protective effect on CD4 counts with either the anti-gp120 aptamer or an aptamer-siRNA combination (that provided more extensive inhibition, resulting in a significantly longer antiviral effect that extended several weeks beyond the last injected dose). [20]
In the discussion after the presentation it was noted that the loss of the patent for the one aptamer on the market has dramatically reduced the cost (from $100,000 down to $25 per mg. However, as they are highly negatively charged this raised the issue of pharmacokinetics and side effects (clotting, platelets and immune effects).
Other subjects covered by the workshop
The workshop explored a wide range of other potential indications for nanomedicines that are not covered in this report including pulmonary fungal and parasite infections (including paracoccidioides brasiliensis), tuberculosis, malaria and prostate cancer.
It also included sessions on regulatory issues (with reference to the recent first meeting of the EMA on nanotechnology) and practical aspects of intellectual property, patents, technology transfer and international research collaborations.
Meeting summary
Many aspects of these new technologies are still in early development, even though some nanoparticle medicines have been used so extensively used that they are now off-patent. Each molecule has specific efficacy and safety issues dependent on the particular manufacturing process and cellular target.
Antiretroviral compounds show exciting pharmacological properties in in vitro studies. Particularly greater potency and that they require lower quantities of the API. The benefits from suspension formulations in development from researchers on nevirapine in South Africa and efavirenz in Argentina could have global impact as paediatric formulations. However, they still need to show proof of concept for overcoming obstacle associated with lymphocyte target cells.
The potential for TB nanoformulations may be closer than those active against HIV. The CSIR researchers have nanoparticle formulations of the four main first-line TB drugs and are working with the TB Alliance on newer compounds including TMC 207.
Some issues from a regulatory safety perspective are still not resolved . The FDA is more advanced than the EMA. Nearly all research is preclinical, with animal or in vitro data to support advantages of nanformulations. Particles based on molecules with established safety data should be easier to assess than totally new constructs. But one of the discussions on regulation of generic formulations of Doxil indicated that the complexity of the manufacturing process is likely to require in vivo safety and efficacy data from new studies in every molecule rather than just bioequivalence studies that enabled easier widespread use of oral antiretrovirals.
It is also important to balance expectations with likely realities for this research. Nanoformulations look as if they will offer exciting solutions to some specific currently unmet challenges rather than the potential to revolutionise treatment for every disease indication. Specific targeting of malignant tissue and its supportive vascular structures is revolutionising approaches to treatment of some cancers.
The political aspect of the workshop included the importance of technology being developed in countries where the demand for final medicines exists. With broadly 10% of global research focused on the 90% of global medical need. Less than 2% of patents are held in Africa and intellectual property and patent legislation are determined to benefit companies based in the developed world.
Of note, several recent reviews have summarised research over the last 15 years into nanoformualations of antiretrovirals. [21, 22]. See Figure 4.
This research is generally conducted by small independent groups working without sufficient funding to discover whether the potential of this technology can be realised for HIV care.
Given the potential advantage of providing reduced toxicity, more durable formulations with improved pharmacokinetics (ie targeting macrophages to increase concentrations in lymph tissue and dramatically extending half-life and dosing intervals) and at a reduced cost makes it frustrating that so far none of these formulations has yet progressed to human studies.
| ARV | Process | Aim/target | Study model | Effect | References |
|---|---|---|---|---|---|
| AZT, 3TC(also d4T, delavirdine, saquinavir) | Multiple polymers and solid lipids. | Aim to cross blood brain barrier by endocytosis and/or phagocytosis to release drug intracellularly. | In vivo (cell) | i.e. permeability of AZT increased 8-20 fold and 3TC by 10-18-fold with PBCA. 100% increase across BBB. | Kuo et al.Int J Pharm 2005, 2006, 2007, 2008. |
| lopinavir | Emulsion templated, freeze-dried nanoparticle dispersions | Improve PK, develop PI formulation that doesnt need ritonavir boosting. | In vivo (cell) | Increased cellular uptake vs aqueous fomulation. | Smith D et al. CROI 2011. |
| d4T, AZT, 3TC, efavirenz | Spray dried PCL nanoparticles. | Improve PK, reduce dosing time and toxicity. | Mouse | Sustained release reducing dosing. | CSIR, South Africa |
| saquinavir | Nanoparticles with PEG and gene delivery | Increase PK, controlled release in mucosal tissue | In vivo | Large particles (200-500 nm) able to overcome challenge of mucosal PK. | Lai et al.Adv Drug Deliv Rev 2009. |
| Ritonavir, lopinavir, efavirenz, indinavir | Added to poly-caprolactone polymer in methylene chloride (multiple emulsion solvent) | Improve PK, reduce dosing time and toxicity.Cross blood-brain barrier and macrophage uptake. | In vitro | Drug detected after 28 days in PBMCs vs <2 days with unencapsulated formulation. | Destache et. CROI 2008.BMC Infect Dis, 2009. |
| CCR5 inhibitor (TAK-779) | Gold nanoparticles as a base scaffold. | Restore activity of an inactive CCR5 inhibitor. | In vitro | Proof of principal for drug delivery. | Bowman et al. CROI 2008. |
| AZT | PLA and PLA-PEG blend particles | Increased uptake by phagocytes. | In vitro | Improved phagocyte uptake with PLA. | Mainardes et al.J Pharm Sci 2009. |
| 3TC, efavirenz | Tuftsin dendrimers | Target macrophages, prolong half-life | In vitro | Cellular uptake >20-fold higher vs free drug, prolonged release >140 hours, increased ARV potency at lower concentrations. | Dutta et al.Biophys Acta 2007Eu J Pharma Sci 2008 |
Compiled from Malipeddi and Rohan [21], Govender [23] and others.
Further reading
The following links are included for further reading.
US NIH nanotechnology research. Links to an extensive NIH funded research programme established in 2005.
http://www.nibib.nih.gov/Research/NIHNano
http://www.nih.gov/science/nanotechnology
EMAs First Scientific Workshop on Nanomedicine. September 2010.
Little information is online but a few large video files from some sessions are available to download.
http://vod.ema.europa.eu/100902
Griffiths G et al. Nanobead-based interventions for the treatment and prevention of tuberculosis. Nature Reviews Microbiology 2010 Nov; 8(11): 827-34.
http://www.ncbi.nlm.nih.gov/pubmed/20938454
Gupta U and Jain NK. Non-polymeric nano-carriers in HIV/AIDS drug delivery and targeting. Advanced Drug Delivery Reviews 62 (2010) 478490.
http://www.ncbi.nlm.nih.gov/pubmed/19913579
Stockley P and Bunka DHJ. Aptamers come of age at last. Nature Reviews, August 2006, (PDF download)
http://www.cs.duke.edu/bioComp/references/hanyingAptamer/aptRev2.pdf
Wong HL et al. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Advanced Drug Delivery Reviews 62 (2010) 503517.
http://linkinghub.elsevier.com/retrieve/pii/S0169409X09003615
References
Unless stated otherwise, all references are to the programme and abstracts of the First Workshop on Nanomedicine for Infectious Diseases of Poverty, 2731 March 2011, Magaliesberg, South Africa. The programme and abstract book can be downloaded free from the workshop website.
http://www.csir.co.za/msm/nano_workshop2011/
- Duncan R. Nanomedicine : from research to health care. Plenary lecture.
- Allen T. Nanomedicines: opportunities and challenges.
- Gabizon A. Liposome drug delivery in cancer therapy: from vision to reality.
- Soznik A. The pros and contras of innovation: issues to take advantage of novel technologies in neglected diseases, the HIV case. Programme page 19.
- Chiappetta DA et al. Efavirenz-loaded polymeric micelles for pediatric anti-HIV pharmacotherapy with significantly higher oral bioavailability. Nanomedicine. 2010;5(1):11-23.
http://www.futuremedicine.com/doi/abs/10.2217/nnm.09.90
Full text online at:
http://www.medscape.com/viewarticle/715261 - Chiappetta DA et al. A highly concentrated and taste-improved aqueous formulation of efavirenz for a more appropriate pediatric management of the anti-HIV therapy. Current HIV Research, 2010.
- Chiappetta DA et al. Oral pharmacokinetics of the anti-HIV efavirenz encapsulated within polymeric micelles, Biomaterials 32 (2011) 2379-2387.
http://linkinghub.elsevier.com/retrieve/pii/S0142961210015371 - Soznik A. Nanotechnology contributions to the pharmacotherapy of pediatric HIV: a dual scientific and ethical challenge and a still pending agenda. Editorial. Nanomedicine (2010) 5(6), 833837.
- Chiappetta DA et al. Synergistic encapsulation of the anti-HIV agent efavirenz within mixed poloxamine/poloxamer polymeric micelles. Nanomedicine: Nanotechnology, Biology and Medicine (in press). doi:10.1016/j.nano.2011.01.017.
http://linkinghub.elsevier.com/retrieve/pii/S1549963411000219 - Katata L et al. Spray dried PCL-Efavirenz nanoparticles for improving the current HIV/AIDS treatment. Abstract 9; page 53.
- Katata L et al. Spray dried PCL-efavirenz nanoparticles for improving the current HIV/AIDS treatment. Poster abstract MOPE0031. Link includes ePoster.
http://pag.aids2010.org/Abstracts.aspx?AID=13785 - van der Merwe H et al. Enhanced in vitro delivery of abacavir and lamivudine entrapped in a Pheroid formulation. Poster 23, page 70.
- Grobler A. The Comparative efficacy and pharmacokinetics of selected anti- infective agents in rodents and primates with and without entrapment in Pheroid technology. Oral presentation, abstract page 22.
- Kotze AF et al. Assessment of the in vitro efficacy of selected artemisinin derivatives in combination with the Pheroid drug delivery system. Poster page 50.
- Wiid I et al Increased bioavailability and in vitro efficacy of anti-tuberculosis drugs by the Pheroid delivery system. Poster 4; page 46.
- Nieuwoudt L-M et al. PheroidTM technology enhance bioavailability of antituberculosis drugs in mice. Poster 18; page 68.
- Hayeshi R et al. Evaluation of polymeric nano drug delivery systems for the treatment of TB. Abstract 19, page 67.
- Khati M. The utility of nano-aptamer bioconjugates against infectious and other diseases common in Africa. Abstract page 18.
- Dey AK et al. An aptamer that neutralizes R5 strains of Human Immunodeficiency Virus type 1 blocks gp120-CCR5 Interaction. J. Virol. November 2005 79: 13806-13810.
http://jvi.asm.org/cgi/content/abstract/79/21/13806 - Neff CP et al. An Aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4+ T cell decline in humanized mice. Sci Transl Med 19 January 2011: Vol. 3, Issue 66, p. 66ra6. DOI: 10.1126/scitranslmed.3001581.
http://stm.sciencemag.org/content/3/66/66ra6.abstract - Mallipeddi R and Rowan LC. Progress in antiretroviral drug delivery using nanotechnology. International Journal of Nanomedicine 2010:5 533547. Online open access.
http://www.dovepress.com/progress-in-antiretroviral-drug-delivery-using-nanotechnology-a4896 - Mamo T et al. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine (London). 2010 February; 5(2): 269285. doi: 10.2217/nnm.10.1.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861897/ - Govender T et al. Polymeric Nanoparticles for Enhancing Antiretroviral Drug Therapy. Drug Delivery, 15:493501, 2008 DOI: 10.1080/10717540802321776.
http://www.ncbi.nlm.nih.gov/pubmed/18720133