
The antiretroviral pipeline
14 July 2011. Related: Pipeline report, Antiretrovirals.
Simon Collins
Introduction
This year the antiretroviral pipeline report is produced against the background of global economic problems that have steadily worsened, with few indications of imminent recovery, the impact of which threatens the goal of universal treatment in both rich and poor countries.
In the United States, the waiting list for State AIDS Drug Assistance Programmes (ADAPs) has increased by 900% in a year to over 8,000 by June 2011. In addition, ADAPs have implemented cost-containment strategies, including reducing formularies, increasing the threshold for financial eligibility, instituting a CD4 threshold of <350 cells/mm3, initiating waiting lists, and capping enrollment and access to the most expensive drugs. This bleak situation for uninsured or underinsured individuals who are otherwise ineligible for government assistance with antiretroviral therapy has so far been managed by patient assistance programmes from drug manufacturers as a response to pressure from US activists in the Fair Pricing Coalition.1, 2
In the UK, the public health care purchasers of HIV services in London (responsible for approximately 50% of HIV-positive people nationally) are seeking to contain national restrictions on National Health Service budgets by cost-saving from the drug budget. From April 2011, two-year contracts have been issued based on each manufacturer tendering bids for bulk volume purchasing. This aims to reduce use of antiretrovirals (ARVs) that have similar efficacy but are significantly more expensive, although treatment remains individualised and all licensed drugs can still be used based on clinical need. This policy was driven by the withdrawal of inflation linked funding and the need to find savings of £8-10 million.3
And last year a challenge to global access, in what was an otherwise hopeful and encouraging pipeline report, came with the first news that donors were restricting access to treatment for new patients in several countries.4 Partly as a result of activist pressure enrollment caps were later removed from PEPFAR programmes in Uganda.66 Constricted health budgets in richer countries now politicise the choice between funding altruistic policies on global heath over those of providing health care for citizens at home.
Of course, in real terms, people in poorer countries are threatened most; they make up a broadly more marginalised demographic that is rarely prioritised for medical services. Despite this, right-wing health economists chose to propose that HIV funding has had a negative impact by dominating sponsor funding.5, 6 In reality, the mobilization to focus on HIV, tuberculosis, and malaria has not only improved health investment in poor countries but has developed and strengthened the health infrastructure that was initially used as an excuse not to provide HIV treatment. Fortunately, each reactionary publication is vigorously rebutted with other articles and letters arguing the importance of the Global Fund to Fight AIDS, Tuberculosis and Malaria as a model for global health interventions.7
Consequently, new strategies are being formulated to try to prevent the potential health disaster that threatens to reverse the achievements of decades of prevention programmes. In May 2011, the results from the HPTN 052 study added to accumulating data that quantify the significant impact of HIV treatment on reducing transmission – by 96% in people starting treatment with CD4 counts >250 cells/mm3.8,9 If the incentive of access to antiretroviral treatment is removed this will reduce the primary incentive for people to test and will drive HIV back underground.
Six million people now on treatment are returned to health, often starting from advanced HIV infection (CD4 counts <100 cells/mm3).10 However, less than 50% of people in need of treatment globally based on criteria of a CD4 count of <200 cells/mm3 and 35% based on 350 cells/mm3 currently access treatment. Additionally, at least half of the people on treatment are using drugs whose greater toxicity increases the risk of serious health complications. The World Health Organization no longer recommends stavudine (d4T) and the European Medicines Agency reassessed the use of stavudine in Europe to circumstances where its use was driven as a lifesaving emergency (specifically based on economic rather than health advantages),11 yet d4T remains one of the most widely used nucleosides in antiretroviral combinations in poor countries. When data on side effects are collected, rates as high as 30-50% for irreversible peripheral neuropathy or lipoatrophy are commonly reported.
The demand for newer and more effective, efficient, safe, and affordable global treatment has never been greater. Western countries will remain a financially profitable market, but this is increasingly dependent on competitive rather than premium pricing.
This report summarises results on pipeline compounds with promising future potential that have been presented in posters and presentations at key conferences. It is dependent on data that has passed some level of peer review, tempering the forward-looking statements of company press releases. It is limited to compounds that have cleared preclinical development and that have in vivo data in HIV-positive people.
Update from the 2010 pipeline report
Since last year’s report, the only new drug to be approved is the NNRTI rilpivirine (Edurant) with a US Food and Drug Administration (FDA) indication for treatment-naive patients in May 2011.12 An extended release formulation of nevirapine was also approved in March 2011.13
Rilpivirine is also coformulated in a fixed-dose combination with tenofovir/FTC that was submitted for approval in November 2010. Although the FDA required additional data, approval is expected, perhaps while this publication is in press.14 Gilead has completed and released 48-week data from ongoing phase III studies of a four-drug fixed dose combination (elvitegravir, cobicistat, tenofovir, and FTC), as it has with studies of the integrase inhibitor elvitegravir and the pharmacokinetic booster cobicistat. Additional pharmacokinetic and drug interaction studies have been reported for both compounds.16, 18
Results from phase II studies of the integrase inhibitor dolutegravir (formerly S/GSK572) in treatment-experienced patients were presented at the 18th Conference on Retroviruses and Opportunistic Infections (CROI) in February 2011, and planned phase III studies in treatment-experienced patients are now open.19
Phase II studies continue or are expected for an attachment inhibitor (BMS-663068), an NNRTI (lersivirine), and a CCR5 inhibitor (cenicriviroc, formerly TBR-652).
However, five compounds listed in last year’s report as being in phase I studies have not visibly progressed, while three compounds had their development put on hold or discontinued: vicriviroc (a CCR5 inhibitor), GSK-761 (an NNRTI), and beviramat (a maturation inhibitor).
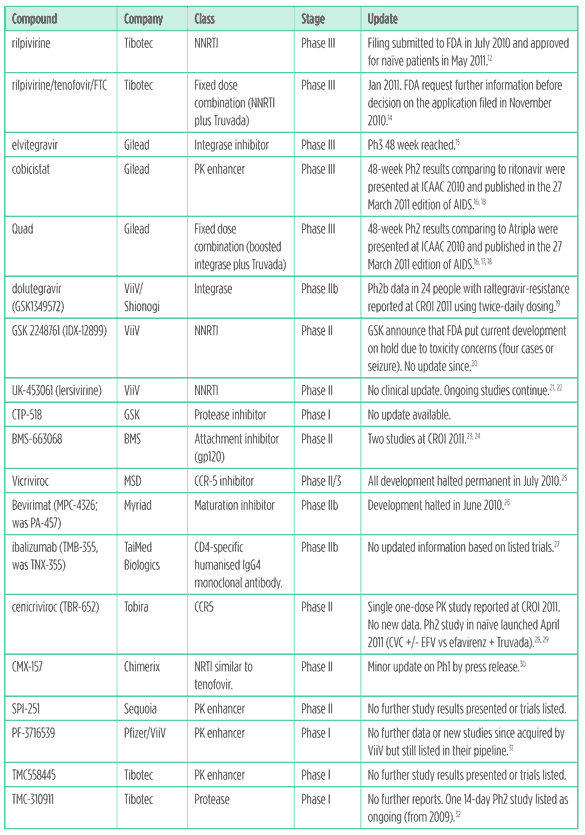
Table 1 summarises developments for the most important compounds highlighted in the 2010 pipeline report.
Table 1: Update on pipeline compounds in 2010 report
New compounds first reported in the last year
Several compounds first reported in vivo results this year and are summarised in Table 2.
GS-7340: a prodrug of tenofovir
GS-7340 is a formulation of tenofovir in development by Gilead that achieves higher levels of the active metabolite in lymph tissue and target cells including peripheral blood mononuclear cells (PBMCs) and has higher potency compared to equivalent tenofovir doses while maintaining reduced plasma concentrations (approximately 100-fold lower). The EC50 of GS 7340 against HIV-1 in MT-2 cells is 0.005 uM compared to 5 uM for tenofovir. This has the potential to require less active pharmaceutical ingredient (API), increase antiviral activity compared to tenofovir, and reduce systemic-related toxicity.
Results from the first dose-ranging study were presented at CROI 2011.33 The double-blind study randomised 30 treatment-naive patients (CD4 >200; viral load >15,000) to either 50 mg or 150 mg of GS-7340 or to tenofovir 300mg (ratio 1:1:1). After 14 days these three groups produced time weighted viral load reductions of -0.95 (+0.32), -1.07 (+0.14) and -0.54 (+0.32) log10 copies/mL, respectively. Mean viral load levels dropped by -0.95, -1.57, and -1.74 log in the tenofovir, 50 mg, and 150 mg arms, respectively. Blood levels were lower (Cmax/AUC by 94%/88% with 50 mg and by 80%/58% with 150 mg) than the tenofovir group with PBMC levels approximately 30-fold higher.
There were no study discontinuations and no grade 3 or 4 events. Side effects reported were generally mild (nausea, headache).
The potential of this compound looks promising. It is unfortunate that it was not prioritised for faster development. In vivo data were presented on GS-7340 nine years ago at CROI 2002.34
Festinavir: an NRTI (previously OBP-601)
Festinavir is an NRTI with a chemical structure similar to stavudine (d4T), but which initial studies indicate should not have the same side effect concerns as it is a weak inhibitor of DNA synthesis in cell studies. BMS acquired development and marketing rights to Festinavir from Oncolys BioPharma in December 2010.35
Results from a phase Ib?IIa dose escalation study were presented as a poster at the 50th ICAAC in September 2010.36
Festinavir monotherapy was given for ten days to four groups of eight treatment-experienced patients currently not on treatment (each 6 active: 2 placebo) using once-daily doses of 100, 200, 300, and 600 mg.
Mean reductions in viral load at day 10 were 0.87, 0.98, 1.36 and 1.22 log10/copies/mL in the 100, 200, 300, and 400mg groups, respectively (vs -0.07 in the placebo group) from baseline levels that ranged from 4.2 – 4.6 log10 copies/mL.
No pattern of side effects appeared over 10 days with all grade 2-3 (n = 13) and grade 4 (n = 2) side effects judged unrelated to the study drug. No new reverse transcriptase mutations emerged at day 10 and 17.
In vitro data on the drug susceptibility of festinavir, including to the Q151M NRTI multidrug resistant mutation were presented in a poster at CROI in 2008.37
Although antiviral activity was reduced in presence of in most viruses carrying nucleoside-associated mutations (5- to 10-fold), including M41L (0.3 to 4.3-fold), and D67N (1.6- to 7.8-fold) resistance mutations, together with K103N +/- M184V. Viruses carrying the Q151M mutation were hypersusceptible to OBP-601 (0.1- to 0.2- fold), even in the presence of K65R (0.3- to 1.3-fold).
Table 2: Compounds with first data presented during the previous year

Update on recent approvals and other compounds in development
This section includes a review of other compounds in further development.
Rilpivirine
Rilpivirine was approved in the United States in May 2011 for treatment-naive patients, based on 48-week results from the ECHO and THRIVE phase III studies and safety results from a phase II study out to 192 weeks.38
These studies, while reporting rilpivirine to be statistically non-inferior when compared to efavirenz, indicated less effective viral suppression when baseline viral load was >100,000 copies/mL with a higher risk of resistance in the context of treatment failure, and a greater loss of efficacy at lower than 95% adherence. These disadvantages were balanced by fewer side effects.39, 40
However, FDA approval is as a treatment for people who have not previously used treatment, when study results suggest use for an early switching of people with intolerance to efavirenz. The prescribing information highlights the poorer virological response at high viral load, and higher risk of resistance.
Rilpivirine is a 25mg tablet that needs to be taken once daily with food. Exposure is reduced by approximately 40% when taken fasted compared to with a normal meal or high-fat meal (defined as 533 kcal and 928 kcal, respectively). Exposures were reduced by 50% when taken with only a high-protein drink.
It was developed at a 25 mg dose due to phase II studies showing similar efficacy at 25, 50 and 75 mg and a caution over cardiovascular toxicity (QTc interval) at higher doses (75 and 150 mg). While the low dose offers exciting future potential for the development of coformulations, it results in a low pharmacokinetic threshold for people with poorer drug absorption.
The phase III studies had a similar design apart from use of background nucleosides. ECHO used tenofovir/FTC for all patients, and THRIVE allowed investigator choice. Each study randomised close to 700 treatment-naive patients without NNRTI or NRTI resistance. The primary endpoint was viral load suppression <50 copies/mL at week 48 (ITT-TLOVR analysis) with a lower margin of -12% difference for non-inferiority. Follow-up continues to week 96.
In the pooled analysis, baseline characteristics of the 1,368 patients included approximate median CD4 count 250 cells/mm3 (range 1-1140), median viral load 5 log copies/mL (range 2-7), with just over 25% of patients having a previous AIDS diagnosis. Gender ratio was 75% male: 25% female and mean age 36 years. Racial demographics were roughly 60% white, 24% black, and 12% Asian. Between 7% and 9% of patients were those coinfected with hepatitis B or C. Nucleoside choice in THRIVE was 60% tenofovir/FTC, 30% AZT/3TC, and 10% abacavir/3TC.
At week 48, suppression to <50 copies/mL was achieved in 84% versus 82% patients in the rilpivirine versus efavirenz groups (pooled results difference +1.6; 95%CI -1.7 to +8.8) with the lower bound for the confidence interval significantly above the prespecified limit. CD4 increases were similar at +192 versus + 176 cells/mm3, respectively.
Differences between the arms were more apparent when looking at reasons for treatment failure. In rilpivirine versus efavirenz, respectively, 9% versus 5% reported virological failure and approximately 2% versus 7% discontinued due to side effects. Around 5% patients discontinued from each arm for other reasons.
In the rilpivirine versus efavirenz groups, 5.5% versus 2.6% of people never suppressed to <50 copies/mL and 3.5% versus 2.2% patients who suppressed later rebounded.
No differences in virological response were reported by gender, race, geographical region, or nucleoside backbone. However, by baseline viral load the pooled response rates were 90% versus 84% (difference +6.6: 95%CI +1.6, +11.5) in favor of rilpivirine in the <100,000 group and 77% versus 81% (difference -3.6: 95%CI -9.8, +2.5) in favor of efavirenz in the >100,000 group. The vulnerability of the low dose was also reflected in the adherence analysis. Viral efficacy was similar between arms when adherence rates were reported as >95% and when viral load was low. However, when adherence dropped to less than 90% efficacy rates were lower with rilpivirine at both high and low viral loads.
People whose treatment failed on rilpivirine developed higher rates of both NNRTI-associated (63% vs 54%) and NRTI-associated (68% vs 32%) mutations. Rilpivirine was associated with the E138K mutation, with 90% of these patients showing phenotypic cross-resistance to etravirine, essentially losing the NNRTI class. People experiencing virological failure on efavirenz commonly developed K103N, which should retain sensitivity to etravirine.
Tolerability results favored rilpivirine with comparisons below for rilpivirine versus efavirenz. While >90% of patients in each arm reported at least one side effect, grade 2?4 events related to study drug occurred in 16% versus 31% (p <0.0001) and discontinuations due to toxicity occurred in 3% versus 8% (p = 0.0005). Neurological side effects occurred in 17% versus 38% (p <0.0001), psychiatric side effects in 15% versus 23% (p = 0.0002), abnormal dreams in 8% versus 13% (p = 0.0061) and rash in 3% versus 14% (p <0.0001).
Grade 3-4 laboratory abnormalities occurred in 11% versus 18% patients (p<0.001), with higher rates of elevated ALT (1.5% vs 3.4%, p <0.05) as well as increases in LDL cholesterol (0.7% vs 4.1%, p <0.0001), triglycerides 0.3% vs 2.2%, p <0.001), and total cholesterol (0.1 vs 2.5%, p <0.0001), all favoring rilpivirine.
There was minimal change in mean serum creatinine in both groups, with no grade 3/4 creatinine increases and no discontinuations due to renal side effects or cases of acute renal failure. No difference was seen in changes in QTc interval between the TMC278 and efavirenz groups.
Several posters at CROI 2011 provided greater details on side effects and tolerability, again from pooled analysis of the same studies. While these generally show a broadly better profile compared to efavirenz, most side effects including central nervous system-related events are reduced rather than eliminated: rilpivirine has a similar profile to efavirenz, though just a little lighter. Some of the differences that are highly statistically significant may have limited clinical impact.
Lipid differences statistically favored rilpivirine over efavirenz, with greater increases in total cholesterol (TC), LDL cholesterol (LDLc), and triglycerides (TG) from baseline to week 48 reported with efavirenz compared to no significant changes in the rilpivirine group. No patients, however, discontinued treatment due to lipid changes. Grades 3/4 lipid-related abnormalities were lower with rilpivirine but also generally low in the study as a whole: TC (0.1% vs 3%, p =0.001), LDLc (1% vs 4%, p =0.001), and TG (0.3% vs 2%, p =0.001). These differences did not result in differences between the two groups in TC/HDL cholesterol ratio or cardiovascular risk as measured by the Framingham score.41
There were differences in the impact on vitamin D levels, measured as 25(OH)D (nmol/L). Mean (SD) baseline and week 48 reductions were statistically greater in patients using efavirenz: 61.8 (26.3) and 60.8 (22.8) [mean change -0.6 (17.9, p = 0.57] vs 64.1 (30.2) to 58.6 (26.9) [mean change -6.1 (18.0), p <0.0001]. The percentage of patients defined as severely deficient remained unchanged in the rilpivirine group at around 4.5% but increased from 5.2% to 9.0% (p = 0.03) in the efavirenz group.42
While neurological complications occurred significantly more frequently in the efavirenz arm, they still occurred in a significant proportion or patients using rilpivirine.43
Rilpivirine fixed-dose combination with tenofovir/FTC
A fixed-dose combination of rilpivirine coformulated with tenofovir/FTC has also been submitted for approval. This will be based on safety and efficacy data from the rilpivarine development programme (the ECHO and THRIVE studies detailed above) plus pharmacokinetic equivalence studies comparing the combined formulation to the separate components.44
Alternative once-daily NNRTI-based single pill combinations clearly benefit patient choice but the efficacy results for this FDC suggest this would be most useful as a rapid switch option from Atripla for people with efavirenz-related side effects. Technically this might be considered to be off-label use unless the treatment-naive for the indication given for rilpivirine is interpreted as non-resistant.
Elvitegravir and Quad: fixed-dose integrase-based combination
Clinical data on elvitegravir in treatment-naive patients includes 48-week results comparing two fixed-dose combinations – Quad (elvitegravir/cobicistat/tenofovir/FTC) versus Atripla (efavirenz/tenofovir/FTC) – were presented at ICAAC in September 2010.16, 17, 18 This updated the 24-week (primary endpoint) results presented earlier at CROI 2010, which showed significantly faster viral responses in the integrase group.
Entry criteria included treatment-naive patients with no documented resistance who were HBV/HCV negative. Baseline demographics included a mean age of 35, approximately 90% of participants were Caucasian, baseline CD4 was 389 versus 450 in the Quad versus Atripla groups and 4-6% had an AIDS diagnosis. However, mean baseline viral load was low at <40,000 copies/mL (4.6 log), and only 25% of people had levels >100,000 copies/mL.
Viral response rates at week 48 were the same as at week 24: 90% versus 83% (p = NS) of patients in the Quad versus Atripla groups respectively had an undetectable viral load (<50 copies/mL) at 48 weeks by intent-to-treat, missing = failure analysis. Mean CD4 increases were higher in the Quad arm at +240 versus +162 cells/mm3.
Tolerability results also matched 24-week data: Quad was better tolerated in terms of lack of efavirenz-related side effects (35% vs 57% with any grade 1-4 drug-related adverse event). This was driven by reduced central nervous system toxicity.
A potentially new once-daily single pill integrase FDC will clearly improve patient options, but if approved, access and use in both first and second-line therapy, is likely to be widely dependent on its price being comparable to currently available options.
Cobicistat
The pharmacokinetic booster cobicistat has so far reported similar boosting efficacy and side effects compared to ritonavir, without residual direct antiretroviral activity. Latest clinical data comes from elvitegravir studies (discussed above) and direct-booster comparison to ritonavir, both presented at the 50th ICAAC in September 2010. As with the studies on Quad in the same population, this showed that 24-week results were maintained out to 48 weeks.16, 17, 18
The comparator-booster study randomised treatment-naive patients to atazanavir boosted by either cobicistat (n = 50) or ritonavir (n = 29). At week 48, the percentage of patients with viral load suppressed to <50 copies/mL by intent-to-treat analysis (missing = failure) was similar with 82% in the cobicistat versus 86 % in the ritonavir groups (p = NS).
Changes in mean estimated glomerular filtration rate (eGFR) at week 24 were similar and didn?t develop with longer follow-up. Other grade 1-4 side effects were seen in 36% versus 48%, respectively. Changes in eGFR seen through 24 weeks were stable and similar to that seen in those receiving ritonavir. No additional discontinuations occurred between weeks 24 and 48: five versus three people in cobicistat versus ritonavir, respectively (relating to side effects in two people vs one).
The primary mechanism for boosting works through inhibition of the cytochrome P450 CYP 3A4. A study in HIV-negative volunteers using phenotype probes to investigate non-3A4-mediated interactions reported only weak inhibition of CYP2D6 and concluded that further interaction studies with metabolites of CYP2D6, CYP2B6, and the P-glyoprotein transporter (P-gp) would not be required.45
BMS-663068: an oral entry inhibitor
BMS-663068 (BMS-068) is an entry inhibitor in development from Bristol-Myers Squibb active against the gp120 binding site on the CD4 cell. After oral administration this prodrug rapidly converts to BMS-626529, which reaches steady state after 2-3 days.
Richard Nettles from BMS reported results from a randomised open-label proof-of-concept study using BMS-068 in 50 people who were either antiretroviral treatment-naive (n = 34) or treatment-experienced but off treatment for the previous eight weeks (n = 16). Pharmacodynamic data were presented for 39 patients with an eligible IC50 <0.1 uM.46
The study included five dose combinations using BMS 068 1200mg once-daily and either 600 mg or 1200 mg twice-daily, with and without ritonavir boosting. Baseline demographics included median (range): CD4 432 cells/mm3 (206-921); viral load 4.4 log copies/mL (3.3-6.1); age 42 years (20-70).
After eight days most doses had reduced viral load by 1.6 logs (ranging from -1.22 to -1.78 in the intent-to-treat and -1.59 to -1.77 in the pharmacodynamic analysis). CD4 cell increases ranged from +28 to +106 after eight days. All patients with an eligible IC50 achieved viral load reductions of at least 1 log.
The pharmacokinetic data showed ritonavir to have a relatively modest impact on boosting BMS-068 and plasma levels of BMS-529 remaining 50-fold above median protein adjusted IC90 for twice-daily dosing and 9-fold above with the once-daily arm (with ritonavir).
All adverse events were grade 1 or 2 and were similar in each arm (though there was not a control arm). The most frequent side effects included headache (22/50,44%) and rash (8/50, 16%), mostly mild. There were no drug discontinuations.
Detailed results were also available in a separate poster available online.47
A pharmacokinetic analysis of the dose response rate reported that the baseline EC90 as a marker for drug susceptibility has a stronger correlation to virological response that pharmacokinetic exposure and that EC90 values were wide in the monotherapy study (median 9.6 ng/mL, range 0.33 to >1860).48
Drug levels suggested that ritonavir boosting may not be needed, and phase IIb trials in treatment-experienced patients are planned to start later this year.
Dolutegravir: a new integrase inhibitor
Dolutegravir (previously GSK1349572) is in development by ViiV/Shionogi as a once-daily integrase inhibitor but overcomes resistance to raltegravir with twice-daily dosing. This makes this a key pipeline compound for use in multidrug-resistant patients.
Results from a second open-label phase IIb study of dolutegravir in people with raltegravir resistance were presented at CROI 2011.49
The dose-response rates from the initial use of a 50 mg once-daily dose of dolutegravir supported increasing the dose to 50 mg twice daily for this second cohort of treatment-experienced patients.
Baseline demographics included median (IQR): CD4 202 cells/mm3 (19-384); viral load 4.3 log copies/mL (3.9-4.8); age 47 (33-68); 75% male; duration on raltegravir 29 months (10-63). At baseline the median (range) fold change in susceptibility was >128 (0.8 to >128) to raltegravir and 2.7 (0.9 to 9.5) to dolutegravir. Baseline patterns of integrase-associated mutations were: N155H (n = 6); Y143H (n = 6); Q148+1 (n = 8); Q148+2 (n = 2); mixture (n = 1); other (n = 1).
The 50 mg twice-daily results included 24 people who added dolutegravir to a failing combination for 11 days (and who dropped raltegravir if they were still taking it). To be included in the study people needed to have at least one additional drug that would be active, and this was added to dolutegravir on day 11 when the background combination was optimised, based on resistance test results.
Nearly all patients (23 out of 24) either reduced their viral load to less than 400 copies/ mL or by at least 0.7 logs. The average (mean) drop in viral load at day 11 was -1.76 logs (SD 0.54) for the study as a whole and -1.57 for people with integrase mutations (Q148 + others). This compared to -1.45 logs seen in the initial 50 mg once-daily study.
Safety data were available for a median 96 days (range 30?172) mainly included common grade 1 or 2 gastrointestinal events not related to dolutegravir. Grade 3 laboratory abnormalities were reported in 4 people (17%) with no discontinuations. One participant had two serious events judged unrelated to the study drug (demyelinating polyneuropathy and diabetes mellitus). No grade 4 events were reported.
The 50mg twice-daily dose has now been selected for phase III studies in people who have integrase inhibitor resistance to raltegravir or elvitegravir. Phase III studies are also ongoing in integrase-naive patients.50
Dolutegravir is also being coformulated in a fixed dose combination with GSK/ViiV nucleosides abacavir and 3TC. A phase III study in treatment-naive patients began recruitment in April 2011.51
Lersivirine: an NNRTI
Lersivirine (formally UK-453061) is an NNRTI previously in development at Pfizer and amalgamated into the ViiV antiretroviral portfolio, where development has not been prioritised over GSK’s pipeline compounds.
A review of lersivirine was presented at the 10th International HIV Congress held in Glasgow in November 2010 focusing on the resistance profile.52
Viral load reductions of 1.6-1.8 logs were previously reported at the three higher doses of a 7 day monotherapy study in treatment-naive patients.
Lersivirine retained in vitro susceptibility (defined as <10-fold change in the IC50 of lersivirine) to 11 out of 19 viruses with multiple NNRTI resistance mutations that had significant loss of sensitivity to etravirine (>2.9-fold change; the lower clinical cutoff for etravirine) and to 5 out of 10 viruses (with >10-fold increase in IC50 for etravirine). Sensitivity to lersivirine was retained for many of the multiple mutations including Y181C. Additionally, the lack of correlation between resistance patterns for lersivirine and etravirine was reported to be consistent with their distinct mechanisms of action.
Cenicriviroc: CCR5 and CCR2 inhibitor (formerly TBR-652 and TAK-652)
Cenicriviroc is a CCR5 inhibitor with CCR2 activity in development with Tobira. CCR2 is associated with and studied in association with diseases related to immune activation.
Results from a 10 day dose-ranging monotherapy study in 54 treatment-experienced but CCR5-receptor blocker-naive patients were presented at the 18th International AIDS Society Conference in July 2010. Participants were randomised to 25, 50, 75, 100, or 150 mg cenicriviroc, all once daily, or to a placebo group. Inflammatory markers (MCP-1, hsCRP, and IL-6) were measured at days 1 and 10.53
Baseline median viral load was 4.5 log copies/mL (range 3.1-6.0), approximately 30,000 copies/mL, but this presumably limited the ability to detect maximum changes for patents starting with low viremia.
At day 10, viral load reductions of 1.4-1.8 log copies/mLwere seen in the 50?150 mg groups. Side effects were generally mild but were dose-related, and were higher in the 100 and 150 mg groups.
Although MCP-1 (a cytokine involved in immune inflammation) increased in all groups except placebo (significantly compared to placebo in the 50, 100, and 150 mg groups) this was only markedly higher for the 150 mg arm (by approximately 350 pg/mL).
Phase IIb studies of the compound are expected to start early in 2011.20
Other research
Limited data available for other pipeline compounds are worth noting.
Apricitabine (ATC), an NRTI with a potential role in multiple drug resistance included in previous TAG reports was reinstated as a compound in development by Avexa, though no new data have been published.54
VIR-576 is a potential fusion inhibitor that targets gp41 that demonstrated mean antiviral activity of ?1.3 log(10) copies/mL in treatment-naive individuals dosed at 5 grams/day (the highest of three dose studies) in a small phase I study. The current formulation, in development by Viro Pharmaceuticals, requires intravenous administration.55
Research continues into modification of antiviral human proteins including APOBEC that are active against HIV, but are neutralised by the accessory HIV viral protein Vif.56, 57, 58 Preclinical studies reporting other potential new compounds that target HIV capsid,Tat inhibitors, RNase H inhibitors, gold-based compounds and numerous other targets are still in preclinical studies.59, 60. 61, 62
The development of new formulations of existing antiretrovirals is an exciting field.
Research-based companies have a long history of reformulating drugs and benefiting from extending patents. Generic formulations and fixed dose combinations have driven access to treatment globally through lower pricing for bulk purchasing and a wider choice of combinations.
For over a decade, generally small groups of scientists have developed numerous nanoformulations of current drugs.63, 64
This wide-ranging technology has the potential to improve on current formulation in many ways, including:
- Better bioavailability; as an example, this could be achieved by designing formulations that overcome hydrophobic or hydrophilic properties of individual molecules.
- Reducing drug wastage by overcoming protein binding during oral absorption, where >90% of the active compounds of antiretroviral drugs are cleared by blood filtration through the liver or kidneys before they are able to act on HIV.
- More targeted delivery should reduce the quantity of raw materials needed.This, in turn, has the potential to have the biggest impact on drugs used in resource-limited settings. Even though the drugs are much cheaper in poorer countries, a much higher percentage of the costs is related to APIs.
- Reducing toxicities related to the metabolism of current oral formulations. For example, if a nanoformulation is designed to increase active drug levels inside cells while keeping blood levels low this has the potential to reduce toxicities related to systemic drug levels.
- Sanctuary site penetration by developing formulations that target cells that cross the blood-brain barrier. In a similar way molecules may be designed to use cells to evade drug transporters such as P-gp that limit penetration of other sites.
Nanobased medicines are already used for other disease areas (including HIV-related complications), but despite the promising results in animal and cell line studies, this has not led to in vivo studies for antiretrovirals.
However, as we went to press, a pharmacokinetic safety study in HIV-negative adults of a paediatric nanosolution of efavirenz was due to start enrollment.65 This encapsulation of efavirenz, otherwise poorly water soluble, into polymeric micelles of different poly(ethylene oxide)-poly(propylene oxide) block copolymers significantly improves oral bioavailability and reduces the interindividual variability. This solution has an improved taste, using only excipients approved by the FDA, and requiring less API has the potential to lower production costs.
Conclusion
Over the last two years a tangible policy shift towards finding a cure for HIV has reestablished the goals of a functional or therapeutic cure high on the research agenda. Like much else, this is driven by the sobering financial challenge of maintaining lifelong treatment for millions of people globally. However, despite the optimism for developing compounds that will target latently infected cells or selectively activate this resting pool, or for immune-based treatments that will maintain viral control without the need for antiretroviral drugs, an HIV cure seems unlikely to be fully realised within ten years.
New treatments will therefore remain in the management of HIV for the foreseeable future, and compounds highlighted in this review will hopefully progress to become licensed medicines. The HIV market in developed countries is continuing to increase annually and treatment in poor countries remains disturbingly less than universal. When approved, the cost of new drugs will drive most aspects of access in all countries.
New pathways still need to be constructed with regulatory support for developing drug options for people with multidrug resistance (MDR), including resistance to integrase inhibitors.
The potential to use an orphan drug designation is only one part of a solution. Rapid access to multiple investigational compounds, likely to be from different companies, is just as essential in order to protect against early failure in the population that is most vulnerable and most dependent on this research.
The requirements for a drug with MDR indication are different than one for use in treatment-naive patients or after early treatment failure because of the different risk-to-benefit ratio on viral efficacy compared to long-term tolerability.
Funding and resources need to be invested in technologies such as nanoformulations that have the potential to really treat HIV universally. This is especially important given the increasing data supporting medical benefits from starting treatment earlier in infection and the additional dramatic impact this has on onward transmission – and the wide gaps yet to be bridged to universal treatment.
References
CROI: Conference on Retroviruses and Opportunistic Infections.
IAS: International AIDS Society
ICAAC: Interscience Conference on Antimicrobial Agents and Chemotherapy
- Based on the US AIDS Drug Assistance Programme (ADAP) waiting list April 2010-April 2011.
http://www.nastad.org - Fair Pricing Coalition brokers rescue of troubled ADAP; nearly 6,500 Floridians will continue to receive HIV medications. Fair Pricing Coalition press statement. (1 February 2011).
http://fairpricingcoalition.org/2011/02/01/fair-pricing-coalition-brokers-rescue-of-troubled-adap-nearly-6500-floridians-will-continue-to-receive-hiv-medications - London HIV Consortium issues new guidelines for ARV prescribing. HIV Treatment Bulletin. March/April 2011.
https://i-base.info/htb/14803 - US government leading backlash against AIDS funding.
http://blog.soros.org/2010/04/u-s-government-leading-backlash-against-aids-funding - Emanuel Z. The HIV/AIDS fight needs cooperation, not division. Huffington Post. 21 July 2010.
http://www.huffingtonpost.com/ zeke-emanuel/aids-activism_b_654710.html - Bongaarts J, Mead O. Global HIV/AIDS policy in transition. Science, Vol. 328 no. 5984 pp. 1359-1360. (1 June 2010).
http://www.sciencemag.org/content/328/5984/1359.short - Sachs J. The MDG decade: looking back and conditional optimism for 2015. Lancet 2010; 376: 950-951.
http://www.thelancet.com/journals/lancet/article/PIIS0140-6736%2810%2961440-7/fulltext - HPTN press release. Initiation of antiretroviral treatment protects uninfected sexual partners from HIV infection (HPTN Study 052): 96% reduction in HIV transmission, according to study conducted by HIV Prevention Trials Network. 12 May 2011.
http://www.hptn.org/web%20documents/PressReleases/HPTN052PressReleaseFINAL5_12_118am.pdf - NIAID. Q&A: The HPTN 052 Study: Preventing Sexual Transmission of HIV with ANTI-HIV Drugs.
http://www.niaid.nih.gov/news/QA/Pages/HPTN052qa.aspx - UNAIDS annual portrait of the global AIDS epidemic. November 2010.
http://www.unaids.org/globalreport/ - EMA issue restricted indication for d4T (stavudine). Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 14-17 February 2011. Questions and answer document:
http://www.ema.europa.eu/ema/pages/includes/document/ open_document.jsp?webContentId=WC500102227 - Approval of Edurant (rilpivirine) a new NNRTI) for the treatment of HIV in treatment naive patients. 20 May 2011.
http://www.fda.gov/ForConsumers/ByAudience/ForPatientAdvocates/HIVandAIDSActivities/ucm256151.htm - Approval of Viramune XR (nevirapine) 400 mg extended release tablet. FDA list serve. (25 March 2011).
http://www.fda.gov/ForConsumers/ByAudience/ForPatientAdvocates/HIVandAIDSActivities/ucm248800.htm - Gilead press release. Gilead receives Refuse to File notification from US FDA on new drug application for single-tablet regimen of Truvada and TMC278. (25 January 2011).
http://www.gilead.com/pr_1519760 - Gilead press release. Phase III clinical trial of Gilead’s investigational elvitegravir meets 48-week primary objective. (23 March 2011).
http://www.gilead.com/pr_1542005 - Elion R et al. The single tablet regimen of elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate (QUAD) maintains a high rate of virologic suppression, and cobicistat is an effective pharmacoenhancer through 48 weeks. 50th ICAAC, 12-15 September 2010, Boston. Abstract H-938b.
http://tinyurl.com/6dp8cbj - Gilead press release. Gilead’s single-tablet ?Quad? HIV regimen maintains high viral suppression through 48 weeks in phase II study. (13 September 2011).
http://www.gilead.com/pr_1470367 - Cohen C et al. Randomised, phase II evaluation of two single-tablet regimens elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for the initial treatment of HIV infection. AIDS. 27 March 2011 – Volume 25 – Issue 6 – p F7-F12.
http://journals.lww.com/aidsonline/Abstract/2011/03270/Randomised,_phase_2_evaluation_of_ two.1.aspx - Eron J et al. DTG in subjects with HIV exhibiting RAL resistance: functional monotherapy results of VIKING study cohort II. 18th CROI, 27 February-3 March 2011, Boston. Abstract 151LB. http://www.retroconference.org/2011/Abstracts/42541.htm
- ViiV press release. (11 February 2011).
http://www.natap.org/2011/newsUpdates/021111_04.htm - Clinical trials registry. Lersivirine 48 week study extended.
http://clinicaltrials.gov/ct2/show/NCT01254656?term=NCT01254656& rank=1
http://clinicaltrials.gov/ct2/results?term=Lersivirine - Mori J et al. Lersivirine: a new NNRTI active across HIV-1 subtypes with a unique resistance profile. 10th International Congress on Drug Therapy in HIV Infection, Glasgow, UK. 7-11 November 2010. Oral abstract O_49.
http://www.jiasociety.org/content/13/S4/ O49/abstract - Nettles R et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068: a potentially first-in-class oral HIV attachment inhibitor. 18th CROI, 27 February-3 March 2011, Boston. Oral abstract 49.
http://www.retroconference.org/2011/Abstracts/41942.htm - Nowicka-Sans B et al. Antiviral activity of a new small molecule HIV-1 attachment inhibitor, BMS-626529, the parent of BMS663068. 18th CROI, 27 February-3 March 2011, Boston. Poster 518.
http://www.retroconference.org/2011/Abstracts/41587.htm - Merck press release. Merck decides to stop development of vicriviroc for treatment of HIV. (14 July 2011).
http://www.hivandhepatitis.com/recent/2010/0716_2010_b.htm - Myriad Pharmaceuticals announces intent to focus on oncology portfolio. (8 June 2010).
http://www.marketwatch.com/story/myriad-pharmaceuticals-announces-intent-to-focus-on-oncology-portfolio-2010-06-08. http://www.hivandhepatitis.com/ recent/2010/0611_2010_c.html - ClinicalTrials.gov listings.
http://clinicaltrials.gov/ct2/results?term=ibalizumab+ - Martin D et al. TBR-652 absorption, distribution, metabolism, and excretion profile in rats, dogs, monkeys, and humans. 18th CROI, 27 February-3 March 2011, Boston. Poster abstract 627.
http://www.retroconference.org/2011/Abstracts/40247.htm - Efficacy, safety, and tolerability of cenicriviroc (CVC) in combination with Truvada or Sustiva plus Truvada in HIV 1-infected, antiretroviral treatment-naive, adult patients infected with only CCR5-tropic virus.
http://clinicaltrials.gov/ct2/results?term=TBR-652 - Chimerix’s antiviral CMX157 demonstrates positive phase I results with favorable pharmacokinetics, safety & tolerability: exhibits potent in vitro activity against XMRV and highly resistant HIV. Chimerix press release. (13 December 2010).
http://www.chimerix-inc.com/news-and-resources/news-and-resources-details/chimerixs-antiviral-cmx157-demonstrates-positive-phase-1-clinical-results-w/ - ViiV Healthcare pipeline. (Accessed 26 April 2011).
http://www.viivhealthcare.com/r-and-d/our-pipeline.aspx - A study to determine the antiviral activity of TMC310911 when administered with ritonavir in treatment-naive HIV-1-infected patients. (Accessed 26 April 2011).
http://clinicaltrials.gov/ct2/show/NCT00838162 - Markowitz M et al. GS-7340 demonstrates greater declines in HIV-1 RNA than TDF during 14 days of monotherapy in HIV- 1-infected subjects. 18th CROI, 27 February-3 March 2011, Boston. Oral abstract 152LB.
http://www.retroconference.org/2011/Abstracts/42549.htm - Lee W et al. In vivo and in vitro characterization of GS 7340, an isopropylalaninyl phenyl ester prodrug of tenofovir; selective intracellular activation of GS 7340 leads to preferential distribution in lymphatic tissues. 9th CROI 24?28 February 2002, Seattle. Poster abstract 384.
http://www.retroconference.org/2002/Abstract/13864.htm - Bristol-Myers Squibb and Oncolys BioPharma enter global licensing agreement for investigational HIV compound. BMS press statement. (20 December 2010).
http://www.oncolys.com/en/our_news/01.html
http://www.bms.com/partnering/Pages/news.aspx - L Cotte et al. A phase-Ib/IIa dose-escalation study of OBP-601 (4?-ethynyl-d4T, festinavir) in treatment-experienced, HIV-1- infected patients. 50th ICAAC, 12?15 September 2010, Boston. Abstract H-933.
http://tinyurl.com/6zefyte - Weber J et al. Drug susceptibility profile of OBP-601, a novel NRTI, using a comprehensive panel of NRTI- or NNRTI-resistant viruses. 15th CROI, 3-6 February 2008, Boston. Poster 726b.
http://retroconference.org/2008/Abstracts/33041.htm - Prescribing information for Edurant (rilpivirine) [Tablets] Initial US Approval: 2011.
http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202022s000lbl.pdf - Cohen C et al. Pooled week 48 efficacy and safety results from ECHO and THRIVE, two double-blind, randomised phase III trials comparing TMC278 versus efavirenz in treatment-naive, HIV-1-infected patients. 18th IAS Conference, 18-23 July 2010, Vienna. Oral abstract late breaker THLBB206.
http://pag.aids2010.org/Abstracts.aspx?SID=1990&AID=17525
Webcast: http://pag.aids2010.org/ flash/?pid=113137 - Rimsky L et al. Characterization of the resistance profile of TMC278: 48-week analysis of the phase III studies ECHO and THRIVE. 50th ICAAC, 12-15 September 2010, Boston. Abstract H-1810.
http://tinyurl.com/6bodpfw - Arribas J et al. Lipid profiles of TMC278 and EFV in treatment-naive HIV-1+ patients: pooled week-48 data from the randomised double-blind phase III ECHO and THRIVE trials. Poster abstract 819.
http://www.retroconference.org/2011/Abstracts/41656.htm - Wohl D et al. Change in vitamin D levels smaller and risk of development of severe vitamin D deficiency lower among HIV-1- infected, treatment-naive adults receiving TMC278 compared with EFV: 48-week results from the phase III ECHO trial. Abstract 79LB.
http://www.retroconference.org/2011/Abstracts/42586.htm - Mills A et al. Neurologic and psychiatric safety profile of TMC278 compared with EFV in treatment-naive HIV-1+ patients: ECHO and THRIVE Trials at 48 Weeks. Poster abstract 420.
http://www.retroconference.org/2011/Abstracts/41718.htm - Mathias A et al. Bioequivalence of the co-formulation of emtricitabine/rilpivirine/tenofovir DF. 18th IAS Conference, 18-23 July 2010, Vienna. Oral abstract late breaker LBPE17.
http://pag.aids2010.org/Abstracts.aspx?AID=17780 - German P et al. The effect of cobicistat on cytochrome P450 2D6, 2B6 and P-glycoprotein using phenotypic probes. 12th International Workshop on Clinical Pharmacology of HIV Therapy, 13-15 April 2011, Miami. Oral abstract O_1.
- Nettles R et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068: a potentially first-in-class oral HIV attachment inhibitor. 18th CROI, 27 February-3 March 2011, Boston. Oral abstract 49.
http://www.retroconference.org/2011/Abstracts/41942.htm - Nowicka-Sans B et al. Antiviral Activity of a New Small Molecule HIV-1 Attachment Inhibitor, BMS-626529, the Parent of BMS-663068.18th CROI, 27 February-3 March 2011, Boston. Poster 518.
http://www.retroconference.org/2011/Abstracts/41587.htm - Zhu L et al. Exposure-response analyses of an oral attachment inhibitor BMS-663068 following 8 days of monotherapy in HIV- infected patients. 12th International Workshop on Clinical Pharmacology of HIV Therapy, 13-15 April 2011, Miami. Oral abstract O_8.
- Eron J et al. DTG in subjects with HIV exhibiting RAL resistance: functional monotherapy results of VIKING study cohort II. 18th CROI, 27 February-3 March 2011, Boston. Abstract 151LB.
http://www.retroconference.org/2011/Abstracts/42541.htm - Shionogi-ViiV Healthcare press release. Clinical programme for investigational once-daily HIV integrase inhibitor – phase III treatment-naive and treatment-experienced trials underway for S/GSK1349572 (?572). 21 October 2010.
http://www.viivhealthcare.com/ media-room/press-releases/2010-10-21.aspx - Shionogi-ViiV Healthcare press release. Shionogi-ViiV Healthcare starts phase III trial for ?572-Trii? fixed-dose combination HIV therapy. (3 February 2011).
http://www.viivhealthcare.com/media-room/press-releases/2011-02-03.aspx - Mori J. Lersivirine: a new NNRTI active across HIV-1 subtypes with a unique resistance profile. 10th International HIV Congress, 7-11 November 2010, Glasgow. Abstract and webcast.
http://www.hiv10.com/webcast.asp?webcast=528 - Martin DE et al. TBR-652, a potent dual chemokine receptor 5/chemokine receptor 2 (CCR5/CCR2) antagonist in phase II development for treatment of HIV infection. 18th IAS Conference, 18-23 July 2010, Vienna. Oral abstract MOAB0104.
http://pag.aids2010.org/Abstracts.aspx?SID=631&AID=8023 - Avexa press statement. Avexa and FDA agree path forward for ATC.
http://www.avexa.com.au/news/press_releases_2011/avexa_ and_fda_agree_path_forwa - Forssmann W-G et al. Short-term monotherapy in HIV-infected patients with a virus entry inhibitor against the gp41 fusion peptide. Sci Transl Med 22 December 2010: Vol.2, Issue 63, p.63re3.
http://stm.sciencemag.org/content/2/63/63re3.short - Nathans R et al. Small-molecule inhibition (by RN-18) of HIV-1 Vif. Research letter. Nature Biotechnology 26, 1187 – 1192 (2008).
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2693000 - Albin JS et al. A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J Biol Chem. 2010 Dec 24;285(52):40785- 92.
http://www.ncbi.nlm.nih.gov/pubmed/20971849 - University of Minnesota press release. U of M scientist gets five-year, $10 million grant to direct innovative HIV research programme. (18 April 2011).
http://www1.umn.edu/news/media-contacts/index.html#Jeff%20Falk - Blair WS et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 6(12): e1001220. doi:10.1371/journal.ppat.1001220.
http://www.plospathogens.org/article/info%3Adoi%2F10.1371%2Fjournal.ppat.1001220 - Chamanian M et al, Unique inhibition of HIV-1 reverse transcription by a cyclic peptide designed as a mimic to the TAR RNA binding site in the HIV-1 Tat protein. Frontiers in HIV Drug Development (HIV DART), 7-10 December 2010, Los Cabos, Mexico. Abstract 29.
http://www.informedhorizons.com/hivdart2010/index.html - Lansdon EB et al. Structural and binding analysis of HIV-1 RNase H inhibitors targeting active site metals. Frontiers in HIV Drug Development (HIV DART), 7-10 December 2010, Los Cabos, Mexico. Abstract 32.
http://www.informedhorizons.com/hivdart2010/index.html - Mphahlele MK et al. Inhibition of reverse transcriptase activity by gold-based compounds. Frontiers in HIV Drug Development (HIV DART), 7-10 December 2010, Los Cabos, Mexico. Abstract 52.
http://www.informedhorizons.com/hivdart2010/index.html - Mallipeddi R and Rowan LC. Progress in antiretroviral drug delivery using nanotechnology. International Journal of Nanomedicine 2010:5 533-547. Online open access.
http://www.dovepress.com/progress-in-antiretroviral-drug-delivery-using-nanotechnology-a4896 - Mamo T et al. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine (London). 2010 February; 5(2): 269-285.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861897 - Personal communication, Dr. Alejandro Sosnik, University of Buenos Aires. (June 2011).
- ?PEPFAR makes U-Turn in Uganda. 4/8/10? 4 August 2010.
http://www.cabsa.org.za/content/pepfar-makes-u-turn-uganda-4810