
HIV pipeline 2017: full version
20 July 2017. Related: Special reports, Supplements, Pipeline report, Antiretrovirals.
This is the full version of HIV pipeline 2017. (Download PDF)
• Read the summary version. • Read IAS 2017 update.
 Introduction
Introduction
This has been a lively year for HIV research.
Even though only one new drug was approved (a once-daily version of raltegravir) several key generic approvals (dolutegravir and TDF/FTC) are perhaps just as important.
Three new fixed-dose combinations (FDCs) have been submitted to the US Food and Drug Administration (FDA)/European Medicines Agency (EMA) with expected decisions soon, including one with a new integrase inhibitor (bictegravir).
Early data presented for several new compounds, including from new drug classes that are in early stages of research, are an optimistic sign that HIV is still a potential market for new drugs.
While current ART is safe and effective there are ways it could become better still: formulations with smaller pills, less frequent dosing, long-acting compounds (weekly, monthly, yearly), with lower doses, fewer side effects and drug interactions, stronger resistance profiles – and it could be cheaper and more accessible. Some of these factors feature in compounds already filed for regulatory approval.
This year the pipeline includes compounds in current classes (NRTIs, NNRTIs, PIs and integrase inhibitors) and compounds with new targets and mechanisms of action (including a capsid inhibitor and several monoclonal antibodies), see Figure 1. It also includes a two-drug combination submitted with an indication as maintenance therapy (dolutegravir/rilpivirine).
Importantly, compounds from new classes – monoclonal antibodies (mAbs), entry inhibitors, maturation inhibitors and capsid inhibitors – are all expected to work for people with multiple drug resistant (MDR) HIV who are dependent on new drugs.
Some of these drugs have potential to be used in very different ways – as treatment, as part of a cure strategy and for prevention as PrEP.
Figure 1: HIV pipeline 2017: targets in the HIV lifecycle
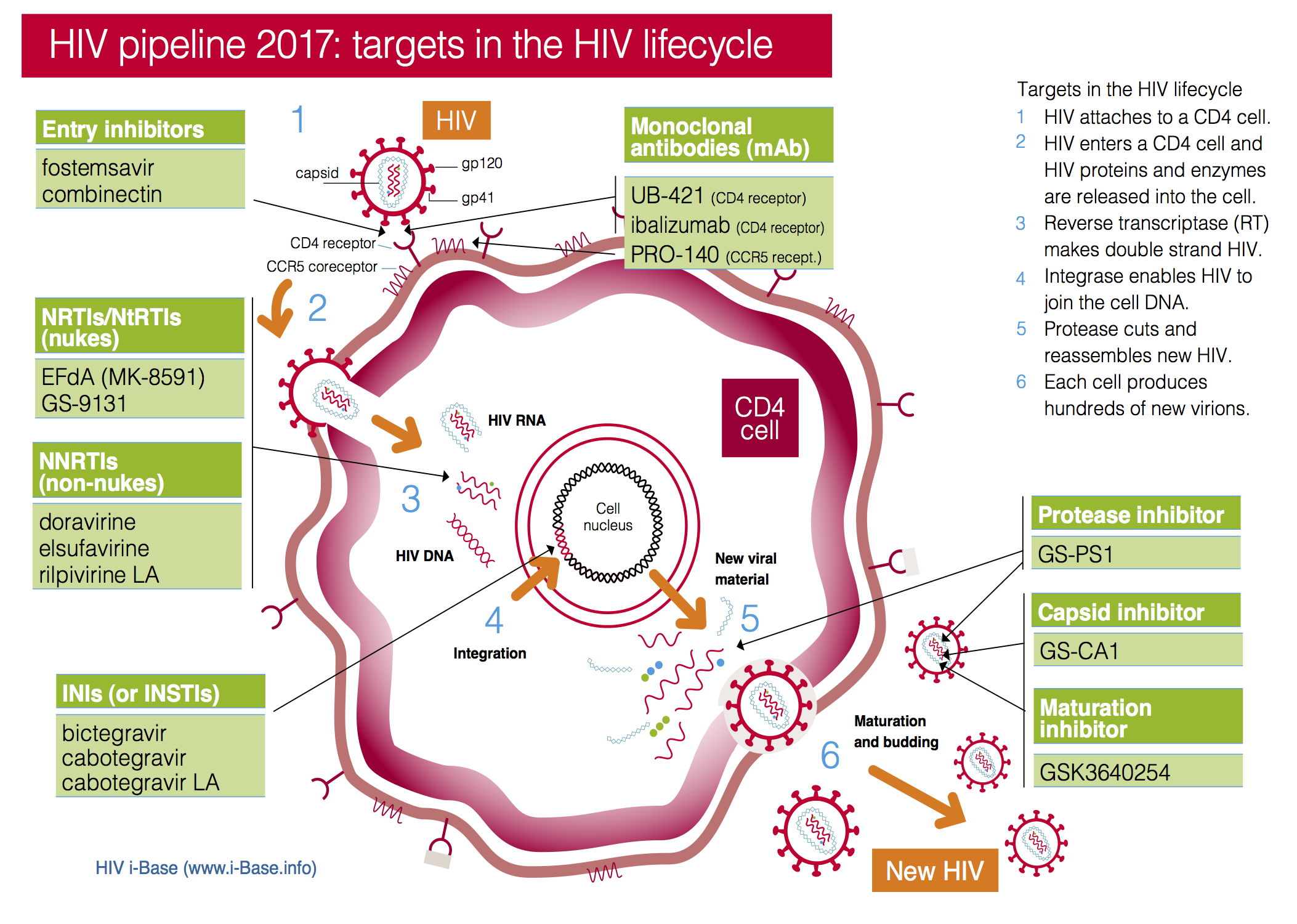
Select image for larger view
Key: INSTI: Integrase strand transfer inhibitors; NRTI: Nucleoside/tide reverse transcriptase inhibitors; NNRTI Non-nucleoside reverse transcriptase inhibitors.
Recently approved new HIV drugs
Raltegravir once-daily
The only new HIV drug approval since the pipeline report in July 2016 was for a once-daily formulation of raltegravir. [1]
Coming nearly ten years after the initial approval of raltegravir, the new version still requires a two-pill dose (2 x 600 mg) but has improved pharmacokinetic (PK) properties that allow once-daily dosing without regard to food, with lower peak drug concentrations and higher trough levels, and less interpatient variability.
Approval was based on 48-week results from the phase 3 treatment-naive ONCEMRK study that similar efficacy and safety to the original formulation: 89% of participants is each arm had undetectable viral load and similar CD4 responses. An updated analysis presented at Glasgow 2016 also showed similar responses with both formulations. [2]
The regulatory approval of several generic formulations over the last year is also important.
Generic dolutegravir
- In September 2016, tentative approval by the FDA of a generic formulation of dolutegravir has the potential to improve treatment options in countries who have access to in-patent generics, especially once the fixed dose combination (with TAF/FTC) also becomes available. [3]
Generic TDF/FTC
- Also in September 2016, in Europe the EMA approved three generic formulations of TDF/FTC [4] that use a different base salt for tenofovir, challenging Gilead’s patent for Truvada. Currently, the UK court has deferred the patent decision to the European Court, making the timeline for access to more affordable versions uncertain as this report went to press. [5]
- The FDA also approved a generic version of TDF/FTC. [6] The patent implications and timeline for US generic access and pricing is unclear and might take years: generic drugs are sometimes priced very highly in the US. [7]
Access to use lower-priced options is essential if PrEP is to have the maximum impact on reducing HIV incidence.
Submitted applications: completed phase 3 results
Several compounds have already been submitted for regulatory evaluation based on primary endpoint results from phase 3 studies.
Darunavir/cobicistat/FTC/TAF – FDC
In September 2016, the first single pill protease-inhibitor based fixed dose combination of darunavir/cobicistat/FTC/TAF (D/C/F/TAF) was submitted in both the US and Europe with a decision expected later in 2017. [8]
The applications are based on results from a randomised, double-blind, active-controlled, phase 3 study in treatment-naive patients with darunavir/cobicistat plus tenofovirDF/FTC as control. [9]
Although these results have not yet been publically presented, the IAS conference will include at least one report from a switching study. [10]
The reduced milligram dose for TAF compared to TDF makes this formulation possible as a single tablet.
Bictegravir/FTC/TAF – FDC
On 12 June 2017, a new drug application was submitted to the US FDA for a single tablet FDC of bictegravir/emtricitabine/tenofovir alafenamide (BIC/FTC/TAF). [11] A similar submission to the EMA in Europe is expected during 3Q 2017.
Bictegravir (formerly GS-9883) is a once-daily integrase inhibitor, that (unlike elvitegravir) does not need to be boosted or to be taken with food. This is also a potent compound, with mean plasma concentration more than 20-fold above the IC95 at 24 hours and low intrapatient differences. Bictegravir is used at low milligram dose (50 mg) leading to a small pill when combined with TAF. Bictegravir has a plasma half-life of 18 hours that suggests some flexibility for adherence and a resistance profile that might retain sensitivity to resistance mutations associated with raltegravir and elvitegravir but that is similar to dolutegravir. [12]
Potential drug-drug interactions that affect bictegravir levels are likely to be dependent on inhibition or induction of both CYP3A4 and UGT1-A1, with little expected effect of bictegravir on levels of other drugs.
The application is based on non-inferiority results from four phase 3 studies. These include a treatment naive study compared to dolutegravir and several switch studies in people with viral suppression on current treatment.
The most recent publicly presented data were results from a phase 2 non-inferiority study compared to dolutegravir that were presented at CROI 2017. [13]
This was a double-blind, placebo controlled, 48-week study that randomised 98 treatment-naive participants 2:1 to either bictegravir 75 mg (n=75) or dolutegravir 50 mg (n=33), both taken once daily with FTC/TAF as background NRTIs. This was a non-inferiority study with the primary endpoint of viral suppression at week 24 and secondary efficacy and safety endpoints at week 48.
Baseline characteristics were similar in each arm and included >90% male, 55% white, median age 30 to 36 years (range 19 to 68). In the bictegravir vs dolutegravir arms, median viral load was 4.41 vs 4.48 log, with 15 vs 21 people >100,000 copies/mL and median CD4 count was 441 vs 455 cells/mm3, with 5 vs 9 participants with CD4 <200 cells/mm3.
At week 24, viral suppression to <50 copies/mL (snapshot analysis) was achieved by 97% vs 94% in the bictegravir vs dolutegravir groups respectively (difference +2.9; 95%CI: –8.5% to +14.2%, p=0.50 NS). For the secondary endpoint at week 48, these rates were 97% vs 91% (difference: +6.4; 95%CI: –6.0% to 18.8%, p=0.17 NS), with no statistical difference between groups. Two participants discontinued from each arm, one in each was lost to follow up, with one due to side effects in the bictegravir arm and one due to adherence in dolutegravir arm. There were no discontinuations to lack of viral efficacy and only one person in each arm discontinued for other reasons while viral load was above 50 copies/mL. Median CD4 increases were similar in each arm: 258 vs 192 cells/mm3 (p=0.16).
Tolerability in both groups was also good with no discontinuations due to treatment-related side effects or deaths – and no significant other differences between groups – though the study numbers were small.
The most common side effects were diarrhoea (12% in each group) and nausea (8% vs 12%). One participant in the bictegravir group discontinued due to urticaria (hives) after week 24. Grade 2 to 4 lab abnormalities including creatinine kinase, AST, hyperglycemia, ALT, LDL, amylase, haematuria, and gylcosuria occurred in 2% to 13% of participants but were generally similar in each group. Small reductions in eGFR (reported with integrase inhibitors) were also similar.
Results are keenly awaited from two phase 3 studies in treatment naive adults that are due to be presented as late breaker abstracts at the IAS 2017 in Paris. [14, 15]
Both studies compare head-to-head the single tablet bictegravir/FTC/TAF FDC to current preferred first-line combinations. One study uses the ViiV FDC of dolutegravir/abacavir/3TC in the comparison arm and the other uses dolutegravir plus TDF/FTC. The results will determine both future use and drug pricing.
Dolutegravir/rilpivirine: two-drug FDC
Although both dolutegravir and rilpivirine are long-approved as oral drugs (in 2013 and 2011 respectively, in the US), in June 2017 a new oral coformulation with both drugs in a single pill was submitted for regulatory approval as an FDC for maintenance therapy. [16]
The application is for use as a switch option in people with suppressed viral load on earlier treatment and is notable for being the first FDC that doesn’t include NRTIs. The application is based on results from the SWORD 1 and 2 studies that were presented as late-breaker abstracts at CROI 2017. [17]
Combined results were for 1024 participants who had undetectable viral load for >12 months and no history of treatment failure who were randomised to switch to either dolutegravir/rilpivirine or continue current ART.
In all analyses at the week-48 primary endpoint, including suppression to <50 copies/mL (ITT: 95% vs. 95%; difference: –0.4%; 95%CI: –3.1% to +2.3%), switching to dual arm was non-inferior to continuing ART.
Dolutegravir-based dual therapy with 3TC is also discussed below.
| Compound/Company | Class | Comment | Refs. | |
|---|---|---|---|---|
| Submitted to FDA/EMA | ||||
| darunavir/cobicistat/ TAF/FTC FDC Janssen and Gilead |
Boosted PI and NRTI FDC | As phase 3 research used darunavir/cobicistat plus TDF/FTC as a comparitor, the FDC is expected to be at least non-inferior. | 8, 9 | |
| bictegravir Gilead |
INSTI and NRTI FDC | Once-daily, unboosted, low mg FDC with FTC/TAF. | 10, 11, 12, 13, 14, 15 | |
| dolutegravir/rilpivirine FDC ViiV and Janssen |
INSTI and NNRTI FDC | Already approved integrase inhibitor but now in a new two-drug coformulation with the NRTI rilpivirine. | 16, 17 | |
| Phase 3 | ||||
| doravirine Merck/MSD |
NNRTI | Active against first generation NNRTI resistance. Non-inferior to efavirenz. Generic FDC with TDF/3TC in phase 3 studies. | 18, 19, 20, 21, 22 | |
| cabotegravir ViiV Healthcare |
INSTI | Oral formulation integrase inhibitor mainly used for lead-in dose before long-acting formulation. | 23 | |
| cabotegravir/rilpivirine LA ViiV Healthcare |
INSTI | Injection with very long half-life – detectable after more than one year following single injection. Research as both treatment in coformulation with rilpivirine LA and prevention as single compound. | 24, 25, 26, 27, 28 | |
| dolutegravir/3TC ViiV |
INSTI and NRTI FDC | Dual combination currently in Phase 3 studies as initial ART in naive participants and a switch option in people on stable ART. | 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 | |
| Ibalizumab TaiMed and Theratechnologies |
mAb
CD4 binding |
Intravenous infusion (800 mg every two weeks) being studied in addition to optimised ART in single arm study in people with multiclass HIV drug resistance. | 40, 41, 42, 43, 44, 45, 46 | |
| PRO 140 CytoDyn |
mAb
CCR5 target |
Once-weekly (350 mg) sub-cutaneous injection with potential to maintain viral suppression for more than two years after stopping ART. Also, with ART against multiclass resistance. | 47, 48, 49 | |
| GSK3684934 ViiV |
attachment inhibitor | Fostemsavir is a gp120 attachment inhibitor that is mainly being studied in treatment-experienced patients with MDR HIV in a large international study. | 50, 51, 52, 53 | |
| Albuvirtide Frontier Biotech |
Entry inhibitor | Similar to T-20 (enfuvirtide) but only requiring once-weekly infusion. Only in development for use in China. | 77 | |
| Phase 2 | ||||
| UB-421 United BioPharma |
mAb
CD4 binding |
Infusion dosed either weekly or every two weeks as alternative to ART during treatment interruption. | 54, 55, 56, 57, 63 | |
| VRC01 | mAb
CD4 binding |
Intravenous infusion (40 mg/kg) being studied with ART for effect on reservoir and in cure research and as PrEP (2 large phase 3 studies are ongoing). Sub-cutaneous dosing of infants to prevent transmission at birth or from breastfeeding. | 57, 58, 59, 60, 61, 62, 63 | |
| elsulfavirine (prodrug of VM-1500A) Viriom |
NNRTI | NNRTI that is being developed for use in low and middle income countries. Similar activity to efavirenz in Russian study. Long-acting formulation being studied with potential for weekly dosing. | 78, 79 | |
| ABX464 Abivax |
Rev inhibitor | Compound with evidence of modest antiviral activity (~0.5 log in 4/6 people) that is also being studied for impact on the viral reservoir. | 81, 82, 83 | |
| Phase 1 and preclinical | ||||
| MK-8591 (EFdA) Merck/MSD |
NRTIs | Highly potent, low dose, active against NRTI resistance. Long half-life, potential as oral (weekly dose) and implant (annual implant for PrEP). | 64, 65, 66, 67 | |
| GS-9131 (prodrug of GS-9141) Gilead |
NRTI | Active against NRTI resistance. Synergy reported with AZT, FTC, abacavir, efavirenz, bictegravir, dolutegravir and lopinavir, and additive activity with TFV and TAF. Will be coformulated with other Gilead drugs. Currently difficult to synthesise in bulk. | 68, 69, 70 | |
| GSK3640254 ViiV Healthcare |
Maturation inhibitor | Back-up compound to first of two maturation inhibitors acquired from BMS. Early preclinical research. | 71, 72, 73 | |
| GSK3732394 ViiV Healthcare |
Entry inhibitor
gp41 and CD4 |
Combined adnectin/fusion inhibitor that stops viral entry by targeting multiple sites of action and the potential for self-administered once-weekly injections. | 74 | |
| GSPI1 Gilead |
Protease inhibitor | New QD unboosted PI, high potency, long half-life, potential in FDC single table regimen (Gilead). | 75 | |
| GS-CA1 Gilead |
capsid inhibitor | Early stage for new class with activity at multiple stages of viral lifecycle. Sub-cutaneous injection with monthly or less frequent dosing. | 76 | |
| Discontinued | ||||
| GS-9695 and GS-9822 Gilead |
INSTI | Non-catalytic integrase inhibitors no longer being studied due to renal toxicity. | 80 | |
| BMS-955176 ViiV Healthcare |
Maturation inhibitor | Development stopped in October 2016 due to problems with GI toxicity and resistance. | 72, 73 | |
Compounds in phase 3 development
The unpredictability of drug development is always important to remember and this year included gains and losses of some compounds and the re-emergence of others.
Doravirine – NNRTI
Doravirine is a once-daily NNRTI that can be taken with or without food that has few drug-drug interactions and that retains activity against common first generation NNRTI mutations (K103N, Y181C, G190A and E138K). Doravirine is being developed in an FDC with generic TDF/3TC and has the compound name MK-1439A. [18]
Results from a two-part, dose-finding, phase 2 study in treatment-naive participants presented at CROI 2016 last year, reported doravirine to be non-inferior compared to efavirenz. [19]
At week 48, viral suppression to <40 copies/mL was reported for 77.8% vs 78.7% of the doravirine vs efavirenz groups respectively, (difference –1.1%; 95%CI –12.2 to +10.0). There were slightly fewer discontinuations in the doravirine group (12% vs 14.7%) and this included fewer discontinuations related to side effects (2.8% vs 5.5%). In patients with baseline viral load <100,000 copies/mL, 87% in each group reached <40 copies/mL at week 48. However, greater viral suppression was reported for the efavirenz group for those starting with viral load >100,000 copies/mL (74% vs 84%). Both groups reported 91% rates using a <200 copy/mL test.
This year at CROI 2017, results from a phase 3 study reported doravirine to be non-inferior compared to boosted darunavir. [20]
This study randomised 769 treatment-naive adults to either doravirine (100 mg) or darunavir/r (800 mg/100 mg), both once-daily with investigator choice of TDF/FTC (87%) or ABC/3TC (13%) as background nukes.
Baseline characteristics include mean age 35 years (SD+/– 10.5), 84% male, 73% white, 22% black with 10% having a clinical history of advanced stage HIV. Mean CD4 and viral load were approximately 420 cells/mm3 (+/– 215) and 4.4 log copies/mL (+/– 0.7 log), with 20% >100,000 copies/mL and 4% >500,00 copies/mL.
At week 48, viral load was <50 copies/mL in 83.8% (321/383) vs 79.9% (306/383), in the doravirine vs darunavir/r arms respectively, (difference 3.9%, 95%CI: –1.6 to +9.4), showing non-inferiority. Results of the stratified analysis of participants with baseline viral load >100,000 copies/mL, were 81.0% (64/79) vs 76.4% (55/72) respectively. Similar suppression was reported for the 17 participants in the doravirine arms with baseline viral load >500,000 copies/mL. CD4 increases were also similar: 193 vs 186 cells/mm3 (difference +7; 95%CI: –21 to +35).
Discontinuations were comparable in each arm although slightly fewer with doravirine than darunavir/r (n=56 (15%) vs n=71 (19%), respectively). Reasons included loss to follow up (4% vs 5%), lack of efficacy 3% vs 4%), withdrawn consent (3% each arm), side effects (1% vs 3%), non-compliance (2% vs 1%), doctor decision (1% in each), pregnancy (n=1 vs 0), protocol violation (<1% vs 2%) and death (n=1 vs 0).
Drug-related side effects were matched between arms. Drug-related side effects were reported in just over 30% of each arm, serious side effects in 5% vs 6% and discontinuations by 1.6% vs 3.1% (doravirine vs darunavir/r respectively).
The most common were diarrhoea (6% vs 13%), nausea (7% vs 8%), and headache (6% vs 3%) for doravirine vs darunavir/r respectively. Fasting LDL-C and non-HDL-C were reduced in the doravirine arm and increased in the darunavir/r arm (–4.5 and –5.3 vs +9.9 and +13.8 mg/dL, p<0.0001).
Drug resistance tests in 7/19 vs 8/24 people who were non-responders or rebounders, included one person with both NRTI and INI resistance at week 24. This person was reported as being non adherent and discontinued at week 24. No PI mutations were observed.
Several new nanoformulations of this compound are also in development. [21]
Results from comparing the M-1439A FDC to efavirenz/TDF/FTC as first-line therapy are due to be presented for the primary endpoint of viral suppression at 48 week in the phase 3 randomised non-inferiority study at the IAS conference in Paris. [22]
Cabotegravir and cabotegravir/rilpivirine long-acting (LA) FDC
Cabotegravir (CAB) is a second-generation integrase inhibitor being developed by ViiV Healthcare as both an oral tablet and long-acting (CAB-LA) injectable formulation. It has potential use as both treatment and, the injectable formulation, as PrEP.
CAB-LA has an extremely long half-life: a single injection results in drug levels that are still detectable in some people more than a year later. This requires that a lead-in phase using the oral formulation is essential before using the injection to screen for likely risk of hypersensitivity reaction. The long half-life means that anyone stopping CAB-LA when used as treatment needs to switch to alternative ART. When used as PrEP, current studies recommend switching to daily oral PrEP for a year.
The oral formulation has a similar drug resistance profile to dolutegravir, and is also being studied as part of dual oral therapy with rilpivirine (see dolutegravir/rilpivirine above), with phase 2b results presented at CROI 2017. [23]
This phase 2b study included 144-week results from 243 treatment-naive participants who started triple therapy ART (dose-ranging cabotegravir or efavirenz, plus background TDF/FTC NRTIs), and who switched to oral cabotegravir plus rilpivirine maintenance therapy at week 24 if viral load was undetectable.
Of 181 participants randomised to CAB, 160 began the dual therapy maintenance phase and 138 entered a further open label phase at week 96.
Although viral suppression was generally good and tolerability included no cabotegravir-related discontinuations for side effects, five patients developed resistance to one or both drugs during the study.
During the maintenance and open label phases, 7 (4%) reported drug-related adverse events (AEs) ≥ Grade 2. Serious AEs occurred in 15 (9%) participants in the CAB group (none drug related) causing 4 people (3%) to withdraw from the study.
Although the CAB entered clinical studies first, phase 3 studies are more advanced for CAB-LA.
Results from the LATTE-2 study, that included 48-week follow-up from using dual injection maintenance therapy (CAB-LA coformulated with rilpivirine LA), were presented at IAS 2016. [24]
This was an open-label phase 2b study in 301 treatment-naive participants, randomised 2:2:1 to 4-weekly (4W) or 8-weekly (8W) injections or to oral ART (cabotegravir plus abacavir/3TC).
The 20-week induction phase used cabotegravir (30 mg once-daily) plus abacavir/3TC once-daily, adding in oral rilpivirine (25 mg once-daily) for the last four weeks. After induction, 91% (n=286) of participants continued into the randomised phase because their viral load was <50 copies/mL. The primary analysis at week 32 of the main study (ie starting after the induction period) was presented at CROI 2016 and these data were used to select the 4-weekly dose for phase 3 studies.
At week 32, viral suppression to <50 copies/mL was achieved in 94%, 95% and 91% of the 4W, 8W and oral arms respectively, meeting pre-specified criteria for showing each CAB-LA arm to not be inferior to oral CAB. Virologic non-response rates were slightly lower in the 4W arm (<1% v 4% in the other arms) with lower non-virologic reasons for discontinuation in the 8W arm.
By week 48, the percentage of participants with viral load <50 copies/mL dropped slightly to 91%, 92% and 89% of the 4W, 8W and oral arms respectively. Virologic non-response was greater in the 8W vs 4W arms (7% vs <1%) but this lead to few discontinuations (<1% vs 0). Discontinuations due to side effects or death was lower in the 8W group (0 vs 5%).
Tolerability at 48 weeks – mainly linked to injection site reactions (ISRs) – was similar to the 32-week results. Slightly higher rates of ISRs in the 8W group levels out to approximately 30% of participants by week 48. Of these, 82% were mild and 17% were moderate: 90% resolved within 7 days. The most common symptoms were pain (67%, nodules (7%) and swelling (6%). Only 2/230 participants (<1%) discontinued due to ISRs.
Other side effects generally occurred at low levels and were similar between injection groups: fever 5% vs 3% vs 0; fatigue 4% vs 2% vs 1%; flu-like symptoms 2% vs 3% vs 0; in the 4W, 8W and oral arms respectively, with headache reported by 2% in all arms.
Virological failure only occurred in two people in the 8W arm and 1 person in the oral group. Mutations associated with drug resistance to integrase inhibitors (Q148R) were only reported in one person in the 8W group.
Also widely reported, participants reported higher rates of satisfaction with injections compared to oral drugs and higher preference for continuing with injecting combination.
Several international phase 3 studies of cabotegravir LA for PrEP are already underway, with oral TDF/FTC as the comparison. [25, 26] New nanoformulations of cabotegravir LA are also in development. [27]
The phase 3 programme includes two large international studies in treatment-naive and -experienced participants: FLAIR (First Long-Acting Injectable Regimen) and ATLAS (Antiretroviral Therapy as Long-Acting Suppression).
Updated 96-week results from LATTE-2 are due to be presented at the IAS 2017 conference in Paris in July. [28]
Dolutegravir/lamivudine FDC
Dolutegravir showed a higher barrier against drug resistance in treatment-naive studies than any other antiretroviral to date and this led to several independent research groups looking at whether dolutegravir could be used in combinations with less than three active drugs.
In addition to using dolutegravir with rilpivirine (see above), several studies are using dolutegravir with lamivudine (3TC) including with the two drugs coformulated in an FDC.
The small single-arm PADDLE study reported rapid reductions in viral load, including in four people with baseline viral load >100,000 copies/mL who maintained undetectable viral load at week-48 in 18/20 treatment-naive participants. [29] Results from week-96 of this study will be presented at IAS 2017. [30]
Several larger phase 2 and 3 studies are ongoing including the single arm LAMIDOL and ACTG A5353 studies and the randomised ASPIRE and TRULIGHT studies. Of these, only the French ANRS 167 LAMIDOL single arm switch study in 104 participants on stable ART has reported results. At CROI 2017, after 40 weeks of dual therapy, 101/104 participants remaining undetectable, with a single person with viral rebound (>50 – 200 copies mL) who switched back to triple ART. [31] However, ACTG A5353 in 122 treatment-naive participants is due to report results at IAS 2017. [32]
The US ASPIRE study and the French TRULIGHT study have randomised 90 and 250 participants respectively who are suppressed on current ART to either switch to dolutegravir/3TC dual therapy or remain on triple ART. [33, 34]
Finally, in August 2016, ViiV announced two large international randomised phase 3 studies (GEMINI 1 and 2) that will compare dolutegravir/3TC FDC to dolutegravir plus separate TDF/FTC. [35] Together these studies will enrol 1400 treatment-naive participants and will quantify whether dual-NRTIs are still needed for some integrase-based regimens, with data collection for the primary endpoint (viral suppression at week-48) expected in 2018. [36, 37]
If these studies produce positive results, a modelling study published last year reported potential savings of $550 million in the US alone over five years if dolutegravir/3TC was used as maintenance therapy by 50% of people who suppressed viral load on triple ART and $800 million if used as initial ART. This increased to $3 billion if 25% of people currently on stable ART switch to dolutegravir/3TC dual therapy. [38]
It is also important that although several studies using dolutegravir as monotherapy maintained viral suppression in most participants, the unpredictable risk of viral rebound in some people with the development of integrase resistance means that monotherapy with dolutegravir is now clearly not recommended. All dolutegravir monotherapy studies should have now changed all participants back to dual or triple therapy. [39]
Ibalizumab – mAb
Ibalizumab is a monoclonal antibody that has been in development for over a decade. Previous development names included TMB-355 and TNX-355 and phase 1 efficacy results were first reported in 2008. [40]
Ibalizumab blocks initial HIV entry by attaching to CD4 receptors and stopping conformational changes that are needed for the virus to enter a CD4 cell. It is active against CCR5 and CXCR4-tropic virus. The half-life of >3 days enables the intravenous (IV) infusion to be given every two weeks.
For much of the development programme, access was limited to an open-label expanded-access study [41] but results from a small phase 3 study (TMB-301) were presented at CROI 2017. [42]
TMB-301 was a single-arm study in 40 participants with multidrug resistant HIV who were failing on their current combination. The study design included adding a single loading 2000 mg dose of ibalizumab to current treatment for two weeks. Background ART was then optimised on day 14 to include at least one sensitive new drug. From day 21 onwards, 800 mg ibalizumab was given every two weeks.
The primary endpoint results are the percentage of participants with >0.5 drop in viral load at day 14.
Mean baseline viral load was 5.0 log copies/mL with 18% > 5 log copies/mL. Mean CD4 was 150 cells/mm3 with 17 people <50 cells/mm3 and 10 people with CD4 between 50 to 200 cells/mm3. More than half the participants had resistance to three classes, one-third to four classes and 40% used another investigational drug (the gp-120 attachment inhibitor fostemsavir) when ART was optimised at day 14.
One week (at day 7) after the initial loading dose (2000 mg IV) added to current failing therapy, viral load dropped by > 0.5 log copies/mL in 83% of participants and > 1.0 log in 60%.
At week 24, mean viral load decrease from baseline was –1.6 log copies/mL, with 55% and 48% having reductions >1 log and >2 log respectively. Viral load was undetectable (<50 copies/mL) in 43% of participants. Baseline CD4 > 50 cells/mm3 was associated with greater viral load reductions.
There were 9/40 discontinuations, mostly (8/9) in participants with lowest CD4 (<50 cells/mm3). This included four deaths (liver failure, KS, end-stage AIDS and lymphoma), all in the low CD4 group. Three people withdrew consent and two were lost to follow-up.
Side effects (n=17) were mostly mild or moderate, but included one case of IRIS.
No treatment related serious side effects or discontinuations were reported during days 0-14.
Results from an intramuscular formulation were also presented at CROI 2017 but although initial viral load reductions were similar to the IV version, rebound after one week suggests greater vulnerability to drug resistance. [43]
Ibalizumab is being developed by the Taiwanese company TaiMed but marketing and distribution rights for the US and Canada have been sold to Theratechnologies (who market tesamorelin for visceral hypertrophy). A press release from the developing companies reported that FDA had granted a priority review with an expected deadline for submission in January 2018. [44]
A further phase 3 study is also ongoing [45] and updated results in treatment-experienced patients are due to be presented at IAS 2017 in Paris in July. [46]
PRO 140 – mAb
PRO 140 is a humanised IgG4 antibody that blocks HIV entry by binding to CCR5 but is active against maraviroc-resistant virus. PRO 140 has been in development for more than ten years, but that paradoxically has been designated “fast-track” status, for having potential activity against MDR HIV.
The most recent phase 3 data were presented at CROI 2017. [47]
This phase 3 study used weekly dosing of 350 mg self-administered sub-cutaneous injections of PRO 140 monotherapy as a switch strategy in participants on stable oral ART with undetectable viral load, who interrupted ART. This was initially a 12-week study with a three-year extension follow-up for people who maintain viral suppression.
Of 41 participants from the first phase, 16 joined the extension phase. Of these, 1/16 withdrew consent, 5/16 had subsequent viral rebound (two consecutive results >400 copies/mL) and 10/16 have maintained viral suppression with follow-up for longer than two years. Of these, 7/10 had undetectable viral load <1 copy/mL, with others at 4, 10 and 19 copies/mL.
No serious side effects or related discontinuations were reported, including low inject site reactions.
Ongoing phase 3 studies include a monotherapy switch study in 300 participants with viral suppression >48 weeks on ART [48] and in addition to ART as part of salvage combination in 30 participants with multidrug resistance to other classes. [49] No new results are expected at IAS 2017.
Fostemsavir – attachment inhibitor
Fostemsavir (GSK3684934) is an attachment inhibitor that binds to gp120 and prevents conformational changes needed for attachment. It is active against nearly all HIV-1 subtypes, though not sub-type AE or group O and has no in vitro cross resistance to drugs from other classes.
This compound is being developed by ViiV but was previously a BMS compound (BMS-663068).
Most recent results were presented at the Glasgow conference in 2016. [50]
This was a subgroup analysis from a BMS phase 2b randomised dose-ranging study in 251 treatment-experienced participants that used atazanavir/r in the control arm. However, rather than using 2 NRTIs as background drugs, all participants used raltegravir (400 mg twice-daily) plus TDF (once-daily) as the background drugs. After 48 weeks, all participants in the GSK-934 arms were switched to the selected dose of 1200 mg once-daily.
Approximate baseline characteristics included median age 39 years (range 20 to 68), 60% of the participants were male, and ethnicity included 40% white, 30% black, 30% other. Mean pre-treatment viral load was 4.85 log copies/mL (SD +/- 0.9 log) and 44% had viral loads >100,000 copies/mL). Mean CD4 count was 230 cells/mm3 (SD +/- 135 cells/mm3) and 39% had <200 CD4 cells/mm3).
Pooled data was presented for all BMS-068 for all participants at 96 weeks, however 30% of these participants and 40% of the control patients discontinued before week 96.
The 96-week efficacy and safety analysis from this study was also reported at CROI 2016, with 61% vs 53% having viral load <50 copies/mL at week 96.
The new analysis reported similar results for the active vs control arms when looking at subgroups for viral load above/below 100,000 copies/mL and for baseline CD4 above/below 200 cells/mm3, gender, age (above/below 50 years) and race/ethnicity. Similar response rates across the active arms were also seen across the range of baseline susceptibility (especially above/below 1.0 nM).
Although side effects were generally mild and similar between group (grade 1 to 4: 91% vs 98%, grade 3/4: 12% vs 14%), a lower percentage of drug-related side effects occurred for the attachment inhibitor (grade 2 to 4: 8.5% vs 37%) and there were fewer drug-related discontinuations (2.5% vs 10.0%). The single death in the active arms was a gunshot wound.
Ongoing research is in a large international phase 3 study (enrolled, no longer recruiting) in treatment-experienced patients with drug resistance and who are sensitive to only two or fewer drug classes. This study was launched in 2015 with an estimated end date in 2020. [51]
Although no new clinical data are due to be presented at IAS 2017, two drug interactions studies are due to be presented as posters. [52, 53]
Compounds in phase 2 studies
UB-421 – mAb
UB-421 is a broadly neutralising mAb that targets CD4 binding with in vitro data suggest comparable or greater potency compared to other compounds, including VRC01 and 3BNC117. [5]
Most recent data were presented at CROI 2017 from a phase 2 study in 29 virally suppressed participants on ART who used UB-421 monotherapy during an 8-week treatment interruption. UB-421 was given by infusion either 10 mg/kg weekly or 25 mg/kg every two-weeks. [54]
Although there were no cases of viral rebound during the monotherapy phase, viral load rebounded at 35 to 62 days after the last UB-421 dose in five participants who delayed restarting ART. All five later restarted ART and viral load became undetectable.
Two current phase 3 studies in people with MDR HIV are listed but not yet enrolling. [55, 56]
No new data are expected at IAS 2017.
VRC01 – mAb
VRC01 is another broadly neutralising mAb that targets the CD4 binding site that can be given by infusion or sub-cutaneous injection and that is in phase 1/2 development with multiple indications: for treatment, as part of cure research and for prevention.
One study at CROI reported no additional impact on reducing the latently infected viral reservoir from adding VRC01 to ART. [57] Other roles in cure research are ongoing [58].
Another study reported tentatively positive safety results from using a single injection in infants after birth to limit risk of vertical transmission and a potential role of additional injections for breastfed infants. [59]
Two other large international dose-finding, placebo-controlled phase 2 studies using VRC01 as PrEP are already ongoing that allow the option for participants to also use open-label oral TDF/FTC PrEP. [60, 61]
Although new clinical data are expected at IAS 2017, one study using this compound as a strategy in cure research is due to be presented, although results showing little impact on the reservoir after stopping ART were published in November 2016. [62, 63]
ABX464 – Rev inhibitor
ABX464 is a molecule thought to work by blocking the end stages of viral assembly. A phase 2a dose-ranging study presented at CROI 2016 in 80 treatment-naive participants in Thailand reported modest antiviral activity (~0.5 log copies/mL) but only in 4/6 people using the highest 150 mg dose (with no responses in 2/6). [81]
The compound is also being studied for impact on viral reservoir and whether it can limit viral rebound in absence of ART, included a related study due to be presented at IAS 2017 in Paris. [82, 83]
Phase 1 and preclinical compounds
As many companies do not widely publicise pre-clinical work, this section is restricted to a few studies.
MK-8591 (EFdA) – NRTI
MK-8591 is a very interesting NRTI now in phase 1 development by Merck that is notable for high potency (currently using a 10 mg oral daily dose), a long plasma half-life that allows once-weekly oral dosing, a slow-release removable implant that might only require annual dosing and ongoing studies looking at use for both treatment and PrEP.
A poster at CROI 2017 reported that, compared to six HIV-1 isolates (including HIV-1 subtypes A, B, C and D and group O), MK-8591 had even better activity in vitro against HIV-2. This includes being fully active against NRTI mutations K65R and Q151M (although the M184V variant conferred 10-fold resistance). [64]
A second poster reported that EFdA reaches good drug levels in vaginal and rectal tissue – supporting further PrEP studies. [65]
IAS 2017 is expected to include new data for both use as treatment and prevention. [66, 67]
GS-9131 – NRTI
GS-9131 is a prodrug of GS-9148 about with early animal and in vitro drug resistance studies presented ten years ago at CROI 2006. [68]
Other published studies highlight the potential for low risk of toxicity in animal studies and retains in vitro phenotypic sensitivity to broad NRTI resistance including mutations at K65R, L74V and M184V and multiple TAMS. [69]
The poster at CROI 2017 confirmed results from previously published studies into the activity against common NRTI mutations. [70]
The compound has good potency (EC50 = 25-200 nM) with activity against HIV-1 subtypes A, B, C, D, E, F, group O and N (EC50 0.29-113 nM), also against HIV-2. Synergistic activity was reported for GS-9131 in combination with AZT, FTC, abacavir, efavirenz, bictegravir, dolutegravir and lopinavir, and additive activity with TFV and TAF.
Currently, GS-9131 is not easy to synthesise and it will need to overcome manufacturing challenges to become easier and cheaper to make. No new data are expected at IAS 2017.
GSK3640254 – maturation inhibitor
The maturation inhibitor GSK3640254 (previously BMS-986197) is in preclinical stages of development with GSK with a molecule acquired from BMS. [71]
An earlier maturation inhibitor, BMS-955176, also acquired from BMS was discontinued in October 2016. [72] The decision to end early the development programme of BMS-955176 was based on 24-week results from the phase 2b AI468-038 study in treatment naive participants. This was due to gastrointestinal intolerability and treatment-emergent drug resistance. The ongoing studies with BMS-955176 (AI468-038 and AI468-048) ended early.
New data on side effects will be presented at IAS 2017. [73]
Combinectin – adnectin/fusion inhibitor
Combinectin (GSK3732394) is a combined adnectin/fusion inhibitor that stops viral entry by targeting multiple sites of action on gp41 and CD4. This compound has the potential for self-administered once-weekly injections.
This compound was in preclinical development with BMS and was acquired by ViiV in late 2015.
Latest data presented at Glasgow 2016 summarised, in vitro activity and resistance data and virologic data from mouse studies. [74]
GS-PI1 – protease inhibitor
GS-PI1 is a once-daily unboosted protease inhibitor with high potency and a long half-life, and in vitro sensitivity against some second-generation PI resistance, in pre-clinical development by Gilead.
An oral presentation at CROI 2017 reported a high barrier to resistance both after in vitro passaging and against multiple resistance complexes from multiple PI-resistant clinical isolates, and pharmacokinetic data from rat and dog studies. [75]
GS-CA1 – capsid inhibitor
First data was presented on GS-CA1, the first compound in a new class of HIV capsid inhibitors, with a formulation that can be used for slow-release injections. [76]
Capsid is the cone-shaped structural core within the virion that protects HIV RNA and related enzymes. As part of a dynamic process, the capsid protein (p24) first breaks down to release viral contents into the CD4 cell to enable reverse transcription and also needs to reassemble inside new virions as part of the maturation process at the end of the lifecycle.
GS-CA1 acts in both the early and late stages by binding at a site that blocks both disassembly and assembly leading to defective new virions that are non-infectious.
The compound is potent with EC50 in target cells of 60 to 140 pM (compared to 1000 to 19000 for efavirenz, dolutegravir and atazanavir) with activity against drug resistance to current HIV classes. Although population sequencing showed the binding site to be highly conserved, capsid resistance can be generated from in vitro serial passaging.
The investigational compound is currently developed as a subcutaneous injection that in rat studies maintained plasma concentrations nine times above the protein adjusted EC95 ten weeks after a single injection. This suggests monthly or longer dosing intervals in humans.
Compounds developed for low and middle-income markets
Although the following compounds are not being developed for use in high-income counties, they are progressing though clinical research.
Albuvirtide – fusion inhibitor
Albuvirtide is a second-generation fusion inhibitor similar to T-20 (enfuvirtide) that is being developed by Frontier Biotechnologies as an alternative second-line combination in China, where access to oral drugs is limited (no integrase inhibitors, or second-generation NNRTIs or PIs).
Early studies from 2012 reported mean viral load reductions of about 1.0 log copies/mL in treatment naive participants using the 320 mg dose following multiple doses over six weeks.
Differences between albuvirtide compared to T-20 include a plasma half-life of 12-14 days allowing once-weekly intravenous infusion (rather than twice-daily sub-cutaneous injections) and a side effect profile that does not include injection site reactions (ISRs).
Partial interim phase 3 results on this compound were presented in Glasgow in October 2016. [77]
This open-label phase 3 non-inferiority study randomised 389 treatment-experienced participants to a second-line combination of lopinavir/r plus either albuvirtide or WHO-recommended NRTIs. Entry criteria included having viral load >1000 copies/mL on first-line ART.
Baseline characteristics included mean age 40 years (SD +/– 11) and 25% were women. Mean CD4 count and viral load were approximately 240 cells/mm3 (SD +/– 140) and 3.8 log (+/– 1.0 log) copies/mL (with 10% having >100,000 copies/mL). Overall, 80% had at least one major drug resistant mutation, with most having both NNRTI and NRTI mutations. Approximately 75% of participants were using tenofovir at baseline.
This interim analysis included 208/389 participants, with results available for 175/208 participants in the modified ITT analysis: 83 vs 92 at 24 weeks and 50 vs 48 at 48 weeks, in the albuvirtude vs NRTI groups respectively.
Although both groups showed 80% viral suppression at 24 weeks, by week 48 rates were 80% vs 66% in the albuvirtide vs control group respectively (difference 14.4%, 95%CI: –3.0 to 31.9). This showed non-inferiority at both time points.
Tolerability was good and most side effects seemed comparable between groups: about 75% reported side effects (mostly mild diarrhoea) with only 5 vs 3% classified as serious events (only one of which, GI-related in the NRTI group, was drug-related). No ISRs were reported.
Based on these results, albuvirtide has already been submitted for conditional approval in China and there are plans to run additional international studies in other countries next year, especially with other long-acting drugs.
A sub-cutaneous formulation of albuvirtide is also in development that would allow self-injections at home, rather than weekly clinic visits needed in the current version.
Elsulfavirine – NNRTI
Elsulfavirine (a pro drug of VM-1500A) is an NNRTI being developed by Viriom for registration in some middle-income countries.
Results from 48-week were presented at CROI 2017 from a randomised, double-blind phase 2b study conducted in Russia in 120 treatment naive participants. Elsulfavirine 20 mg was compared to efavirenz 600 mg, each with tenofovir-DF/FTC background NRTIs. [78]
The elsulfavirine arm reported similar viral suppression to <50 copies/mL (81% vs 73%), including those with baseline viral load >100,000 copies/mL (78% vs 62%), with fewer CNS side effects (32% vs 62%).
A long-acting injectable formulation is being used in ongoing studies for treatment and PrEP and results will be presented at IAS 2017 in Paris this summer. [79]
Other compounds: trailing or lost
Several other compounds that featured in earlier pipeline reports have not lead to new data being presented over the last year.
GS-9695, GS-9822 – integrase inhibitors
GS-9695 is a non-catalytic integrase inhibitor (NCINI) that binds to a conserved pocket on the enzyme that is also targeted by LEDGF. The compound (previously developed by Boehringer Ingelheim as BI-224436) has potential for low dose, once-daily dosing, unboosted, with high barrier to drug resistance. A follow-on compound GS-9822 strengthened the resistance profile.
However, a poster at CROI 2017 reporting unpredictable kidney/urothelial toxicty in monkeys has led to discontinuation of development of this compound. [80]
BMS-955176 – maturation inhibitor
The decision to end the development programme for BMS-955176 due to gastrointestinal intolerability and treatment-emergent drug resistance was already mentioned earlier in this report. [71]
The follow-on compound GSK3640254 is still in development.
Conclusion
This is still an exciting time for HIV drug development.
This year the HIV pipeline is remarkable for a potential range of drugs that could improve many aspects of the traditional approach to treating HIV using three-drug oral therapy.
It includes responses to the changing situation in which all countries now have access to some generic antiretrovirals – and drug pricing will continue to drive access in all countries. It also includes some compounds that are only developed for low- and middle-income countries, and coformulations that will not be available in high income-countries.
References
Key: CROI: Conference on Retroviruses and Opportunistic Infections; IAS: International AIDS Society; HIV Glasgow: Glasgow Congress on HIV Therapy.
- Merck press statement. Merck receives FDA approval of Isentress HD (raltegravir), a new once-daily option, in combination with other antiretroviral agents, for the treatment of HIV-1 infection in appropriate patients. (30 May 2017).
http://investors.merck.com/news/press-release-details/2017/Merck-Receives-FDA-Approval-of-ISENTRESS-HD-raltegravir-a-New-Once-Daily-Option-in-Combination-with-Other-Antiretroviral-Agents-for-the-Treatment-of-HIV-1-Infection-in-Appropriate-Patients/default.aspx - Cahn P et al. Subgroup analyses from ONCEMRK, a phase 3 study of raltegravir (RAL) 1200 mg once daily versus RAL 400 mg twice daily, in combination with tenofovir/emtricitabine, in treatment-naïve HIV-1-infected subjects. HIV Glasgow, 23-26 October 2016. Oral abstract O334.
https://vimeo.com/189136477 (webcast) - Aurobindo Pharma receives US FDA tentative approval for dolutegravir. 22 September 2016.
http://www.clintonhealthaccess.org/usfda-tentative-approval-dolutegravir - EMA Committee for Medicinal Products for Human Use (CHMP). Emtricitabine/tenofovir disoproxil Zentiva, Summary of opinion (initial authorisation). EMA/CHMP/596525/2016. (15 September 2016).
http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/004137/WC500212887.pdf (PDF) - England and Wales High Court (Patents Court) Decisions. Teva UK Ltd & Ors v Gilead Sciences Inc [2017] EWHC 13 (Pat) (13 January 2017).
http://www.bailii.org/ew/cases/EWHC/Patents/2017/13.html - US Food and Drug Administration. FDA approves generic emtricitabine and tenofovir disoproxil fumarate tablets, 200 mg/300 mg (June 9, 2017)
https://content.govdelivery.com/accounts/USFDA/bulletins/1a0e24c - Salzman S. Surprise FDA approval of generic Truvada is a ‘wakeup call’ for activists. TheBody.com. (6 July 2017).
http://www.thebody.com/content/80139/surprise-fda-approval-of-generic-truvada-is-a-wake.html - Janssen press statement. Janssen submits marketing authorisation application for darunavir-based single tablet regimen for treatment of HIV-1 to European Medicines Agency. (12 September 2016).
http://www.janssen.com/janssen-submits-marketing-authorisation-application-darunavir-based-single-tablet-regimen-treatment - ClinicalTrials.gov. A phase 3, randomized, active-controlled, double-blind study to evaluate efficacy and safety of darunavir/cobicistat/emtricitabine/tenofovir alafenamide (D/C/F/TAF) once daily fixed dose combination regimen versus a regimen consisting of darunavir/cobicistat fixed dose combination coadministered with emtricitabine/tenofovir disoproxil fumarate fixed dose combination in antiretroviral treatment-naive HIV-1 infected subjects. NCT02431247.
https://clinicaltrials.gov/ct2/show/NCT02431247 - Molina JM et al. Efficacy and safety of switching from boosted-protease inhibitor plus emtricitabine/tenofovir disoproxil fumarate regimens to the single-tablet regimen of darunavir/cobicistat/emtricitabine/tenofovir alafenamide (D/C/F/TAF) in virologically-suppressed, HIV-1-infected adults through 24 weeks: EMERALD study. IAS 2017, 23-26 July, Paris. Oral abstract TUAB0101.
http://programme.ias2017.org/Abstract/Abstract/4194 - Gilead press statement. Gilead submits new drug application to U.S. food and drug administration for fixed-dose combination of bictegravir, emtricitabine and tenofovir alafenamide for HIV treatment. (12 June 2017).
http://www.gilead.com/news/press-releases - Tsiang M et al. Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1 integrase strand transfer inhibitor with an improved resistance profile. Antimic Agents and Chem. (September 2016). doi: 10.1128/AAC.01474-16.
http://aac.asm.org/content/early/2016/09/13/AAC.01474-16.abstract - Sax PE et al. Randomized trial of bictegravir or dolutegravir with FTC/TAF for initial HIV therapy. CROI 2017, 13-16 February 2017, Seattle. Oral abstract 41.
http://www.croiconference.org/sessions/randomized-trial-bictegravir-or-dolutegravir-ftctaf-initial-hiv-therapy - Gallant J et al. A phase 3 randomized controlled clinical trial of bictegravir in a fixed dose combination, B/F/TAF, vs ABC/DTG/3TC in treatment-naïve adults at week 48. IAS 2017, 23-26 July, Paris. Late breaker oral abstract MOAB0105LB.
http://programme.ias2017.org/Abstract/Abstract/5783 - Sax PE et al. Phase 3 randomized, controlled clinical trial of bictegravir coformulated with FTC/TAF in a fixed-dose combination (B/F/TAF) vs dolutegravir (DTG) + F/TAF in treatment-naïve HIV-1 positive adults: week 48 results. IAS 2017, 23-26 July, Paris. Late breaker oral abstract MOAB0105LB.
http://programme.ias2017.org/Abstract/Abstract/5793 - ViiV Healthcare press statement. ViiV Healthcare submits regulatory applications for the first HIV maintenance regimen comprising only two medicines. (01 June 2017).
https://www.viivhealthcare.com/media/press-releases/2017/june/viiv-healthcare-submits-regulatory-applications-for-the-first-hiv-maintenance-regimen-comprising-only-two-medicines.aspx - Llibre JM et al. Phase III SWORD 1&2: Switch to DTG+RPV/ maintains virologic suppression through 48 wks. CROI 2017, 13-16 February, Seattle. Late breaker oral abstract 44LB.
http://www.croiconference.org/sessions/phase-iii-sword-12-switch-dtgrpv-maintains-virologic-suppression-through-48-wks - ClinicalTrials.gov listing. Effects of switching from ATRIPLA (efavirenz, tenofovir, emtricitabine) to MK-1439A (doravirine, tenofovir, lamivudine) in virologically-suppressed participants (MK-1439A-028). NCT02652260.
https://clinicaltrials.gov/ct2/show/NCT02652260 - Gatell JM et al. Doravirine 100mg QD vs efavirenz +TDF/FTC in ART-naive HIV+ patients: week 48 results. 23rd CROI, 22 – 25 February 2016, Boston. Poster abstract 470.
http://www.croiconference.org/sessions/doravirine-100mg-qd-vs-efavirenz-tdfftc-art-naive - Molina J-M et al. Doravirine is non-inferior to darunavir/r in phase 3 treatment-naive trial at week 48. CROI 2017, 13-16 February 2017, Seattle. Late breaker oral abstract 45LB.
http://www.croiconference.org/sessions/doravirine-non-inferior-darunavirr-phase-3-treatment-naïve-trial-week-48 - ClinicalTrials.gov [Internet]. A rapid pharmacokinetic trial of the bioavailability of four MK-1439 nano formulations in healthy adults. Identifier NCT02549040.
https://clinicaltrials.gov/ct2/show/NCT02549040 - Squires K et al. Fixed dose combination of doravirine/lamivudine/TDF is non-inferior to efavirenz/emtricitabine/TDF in treatment-naive adults with HIV-1 infection: week 48 results of the Phase 3 DRIVE-AHEAD study. IAS 2017, 23-26 July, Paris. Oral late-breaker abstract TUAB0104LB.
http://programme.ias2017.org/Abstract/Abstract/5585 - Margolis DA et al. Long-term safety and efficacy of CAB and RPV as 2-drug oral maintenance therapy. CROI 2017, 13-16 February, Seattle. Poster abstract 442.
http://www.croiconference.org/sessions/long-term-safety-and-efficacy-cab-and-rpv-2-drug-oral-maintenance-therapy - Margolis D et al. Cabotegravir + rilpivirine as long-acting maintenance therapy: LATTE-2 week 48 results. AIDS 2016, 18-22 July 2016, Durban. Oral late breaker abstract THAB0206LB.
http://programme.aids2016.org/Abstract/Abstract/10517 - ClinicalTrials.gov. Evaluating the safety and efficacy of long-acting injectable cabotegravir compared to daily oral TDF/FTC for pre-exposure prophylaxis in HIV-uninfected women. NCT03164564.
https://clinicaltrials.gov/ct2/show/NCT03164564 - ClinicalTrials.gov. Safety and efficacy study of injectable cabotegravir compared to daily oral tenofovir disoproxil fumarate/emtricitabine (TDF/FTC), for pre-exposure prophylaxis in HIV-uninfected cisgender men and transgender women who have sex with men. NCT02720094.
https://clinicaltrials.gov/ct2/show/NCT02720094 - Zhou T et al. A long-acting nanoformulated cabotegravir prodrug for improved antiretroviral therapy. CROI 2017, 13-16 February, Seattle. Poster abs 439.
http://www.croiconference.org/sessions/long-acting-nanoformulated-cabotegravir-prodrug-improved-antiretroviral-therapy - Eron J et al. Safety and efficacy of long-acting CAB and RPV as two drug IM maintenance therapy: LATTE-2 week 96 results. IAS 2017, Paris. Late breaker oral abstract MOAX0205LB.
http://programme.ias2017.org/Abstract/Abstract/5628 - Cahn P et al. Dolutegravir-lamivudine as initial therapy in HIV-infected, ARV naive patients: 48 week results of the PADDLE trial. AIDS 2016, 18-22 July 2016, Durban. Oral late breaker abstract FRAB0104LB.
http://programme.aids2016.org/Abstract/Abstract/10270
http://www.natap.org/2016/IAC/IAC_92.htm (Slides) - Figueroa MI et al. Dolutegravir-lamivudine as initial therapy in HIV-infected, ARV naive patients: 96 week results of the PADDLE trial. IAS 2017, Paris. Poster abstract MOPEB0287.
http://programme.ias2017.org/Abstract/Abstract/1984 - Joly V et al. Promising results of dolutegravir + lamivudine maintenance in ANRS 167 LAMIDOL trial
. CROI 2017, 13-16 February 2017, Seattle. Poster abstract 458.
http://www.croiconference.org/sessions/promising-results-dolutegravir-lamivudine-maintenance-anrs-167-lamidol-trial - Taiwo BO et al. ACTG A5353: a pilot study of dolutegravir (DTG) + lamivudine (3TC) for initial treatment of HIV-1-infected participants with HIV-1 RNA < 500,000 copies/mL. IAS 2017, Paris. Poster abstract TULBPEB21.
http://programme.ias2017.org/Abstract/Abstract/5634 - ClinicalTrials.gov. Dolutegravir antiretroviral strategy to promote improvement and reduce drug exposure study (ASPIRE). NCT02263326.
https://clinicaltrials.gov/ct2/show/NCT02263326 - ClinicalTrials.gov. Trial to evaluate the interest of a reductive anti retroviral strategy using dual therapy inspite of triple therapy (TRULIGHT). NCT02302547.
https://www.clinicaltrials.gov/ct2/show/NCT02302547 - ViiV press release. ViiV Healthcare launches phase III programme evaluating a two-drug regimen combining dolutegravir and lamivudine for HIV-1 treatment. (16 August 2016).
https://www.viivhealthcare.com/media/press-releases/2016/august/viiv-healthcare-launches-phase-iii-programme-evaluating-a-two-drug-regimen-combining-dolutegravir-and-lamivudine-for-hiv-1-treatment.aspx - ClinicalTrials.gov. An efficacy, safety, and tolerability study comparing dolutegravir plus lamivudine with dolutegravir plus tenofovir/emtricitabine in treatment naïve hiv infected subjects (Gemini 1). NCT02831673.
https://www.clinicaltrials.gov/ct2/show/NCT02831673 - ClinicalTrials.gov. An efficacy, safety, and tolerability study comparing dolutegravir (DTG) plus lamivudine (3TC) with dolutegravir plus tenofovir/emtricitabine in treatment naïve hiv infected subjects (Gemini 2). NCT02831764.
https://www.clinicaltrials.gov/ct2/show/NCT02831764 - Girouard M et al. The cost-effectiveness and budget impact of 2-drug dolutegravir-lamivudine regimens for the treatment of HIV infection in the United States. Clin Infect Dis. 2016 Mar 15; 62(6): 784–791.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4772845 - Collins S. Dolutegravir monotherapy studies halted due to integrase resistance: dual therapy studies continue. HTB, February 2017.
https://i-base.info/htb/31289 - Jacobson JM et al. Safety, pharmacokinetics, and antiretroviral activity of multiple doses of ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in human immunodeficiency virus type 1-infected adults. Antimicrob Agents Chemother. 2009; 53:450-7.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC263062 - ClinicalTrials.gov. Ibalizumab plus optimized background regimen in patient with multi-drug resistant HIV. NCT02475629.
https://clinicaltrials.gov/ct2/show/study/NCT02475629 - Lewis S et al. Long-acting ibalizumab in patients with multi-drug resistant HIV-1: A 24-week study. CROI 2017, 13-16 February, Seattle. Poster abstract 449LB.
http://www.croiconference.org/sessions/long-acting-ibalizumab-patients-multi-drug-resistant-hiv-1-24-week-study - Lin H-H et al. Intramuscular ibalizumab: pharmacokinetics, safety, and efficacy vs IV administration. CROI 2017, 13-16 February, Seattle. Poster abstract 438.
http://www.croiconference.org/sessions/intramuscular-ibalizumab-pharmacokinetics-safety-and-efficacy-vs-iv-administration - Theratechnologies press release. Biologics License Application (BLA) accepted for review with a target action date of January 3, 2018. (30 June 2017).
http://theratech.com/sites/default/files/news_release_en/nr-20170630-en.pdf (PDF) - ClinicalTrials.gov. Ibalizumab plus optimized background regimen in treatment-experienced patients with multi-drug resistant HIV-1. NCT02707861.
https://clinicaltrials.gov/ct2/show/NCT02707861 - Weinheimer S et al. Long-acting ibalizumab susceptibility in multi-drug resistant HIV patients. IAS 2017, Paris. Poster abstract MOPEB0352.
http://programme.ias2017.org/Abstract/Abstract/4685 - Lalezari J et al. PRO140 single-agent maintenance therapy for HIV-1 infection: a 2-year update. CROI 2017, 13-16 February, Seattle. Poster abstract 437.
http://www.croiconference.org/sessions/pro140-single-agent-maintenance-therapy-hiv-1-infection-2-year-update
http://www.croiwebcasts.org/p/2017croi/croi33640 (webcast) - ClinicalTrials.gov. Study of PRO 140 SC as single agent maintenance therapy in virally suppressed subjects with CCR5-tropic HIV-1 infection. NCT02859961.
https://clinicaltrials.gov/ct2/show/NCT02859961 - ClinicalTrials.gov. A randomized, double-blind, placebo-controlled trial, followed by single-arm treatment of PRO 140 in combination w/ optimized background therapy in treatment-experienced HIV subjects (PRO 140). NCT02483078.
https://clinicaltrials.gov/ct2/show/NCT02483078 - Llamoso C et al HIV-1 attachment inhibitor prodrug BMS-663068 in antiretroviral-experienced subjects: week 96 safety analysis. Glasgow Congress 2016. 23-26 October. Late breaker oral abstract O335B.
https://player.vimeo.com/video/189136479 (webcast) - ClinicalTrials.gov. Attachment inhibitor comparison in heavily treatment experienced patients. NCT02362503.
https://clinicaltrials.gov/ct2/show/NCT02362503 - Sevinsky H et al. The effect of fostemsavir on methadone and buprenorphine pharmacokinetics. IAS 2017, Paris. Poster abstract MOPEB0338.
http://programme.ias2017.org/Abstract/Abstract/3300 - Magee M et al. The effect of fostemsavir on the pharmacokinetics of a combined oral contraceptive (OC) containing ethinyl estradiol (EE) and norethindrone (NE) in healthy female subjects. IAS 2017, Paris. Poster abstract MOPEB0339.
http://programme.ias2017.org/Abstract/Abstract/3312 - Wang C-Y et al. A phase 2 open-label trial of antibody UB-421 monotherapy as a substitute for HAART. CROI 2017, 13-16 February, Seattle. Poster abstract 450 LB.
http://www.croiconference.org/sessions/phase-2-open-label-trial-antibody-ub-421-monotherapy-substitute-haart - ClinicalTrials.gov. To investigate the efficacy and safety of UB-421 monotherapy in HIV-1 infected adults. NCT0314921.
https://clinicaltrials.gov/ct2/show/NCT0314921 - ClinicalTrials.gov. UB-421 combine with optimized background therapy regimen in multi-drug resistant HIV-1 infection patients. NCT03164447.
https://clinicaltrials.gov/ct2/show/NCT03164447 - Riddler S et al. VRC01 infusion has no effect on HIV-1 persistence in ART-suppressed chronic infection. CROI 2017, 13-16 February, Seattle. Late breaker poster 330LB.
http://www.croiconference.org/sessions/vrc01-infusion-has-no-effect-hiv-1-persistance-art-suppressed-chronic-infection - Schief WR et al. Immunogen design to induce HIV neutralizing antibodies. CROI 2017, 13-16 February, Seattle. Oral abstract 143.
http://www.croiconference.org/sessions/immunogen-design-induce-hiv-neutralizing-antibodies (abstract and webcast) - Cunningham CK et al. Safety & pharmacokinetics of the monoclonal antibody, VRC01, in HIV-exposed newborns. CROI 2017, 13-16 February, Seattle. Poster abstract 760.
http://www.croiconference.org/sessions/safety-pharmacokinetics-monoclonal-antibody-vrc01-hiv-exposed-newborns - ClinicalTrials.gov [Internet]. Evaluating the safety and efficacy of the VRC01 antibody in reducing acquisition of HIV-1 infection in women. NCT02568215.
https://www.clinicaltrials.gov/ct2/show/NCT02568215 - ClinicalTrials.gov [Internet]. Evaluating the safety and efficacy of the VRC01 antibody in reducing acquisition of HIV-1 infection among men and transgender persons who have sex with men. NCT02716675.
https://www.clinicaltrials.gov/ct2/show/NCT02716675 - Crowell TA et al. HIV-specific broadly-neutralizing monoclonal antibody, VRC01, minimally impacts time to viral rebound following treatment interruption in virologically-suppressed, HIV-infected participants who initiated antiretroviral therapy during acute HIV infection. IAS 2017, Paris. Oral abstract TUAB0106LB.
http://programme.ias2017.org/Abstract/Abstract/5527 - Barr K et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N Engl J Med 2016; 375:2037-2050. DOI: 10.1056/NEJMoa1608243. (24 November 2016).
http://www.nejm.org/doi/full/10.1056/NEJMoa1608243 - Wu V et al. Antiviral activity of EFdA against NRTI-sensitive and resistant strains of HIV-2. CROI 2017, 13-16 February, Seattle, Washington. Poster abstract 440.
http://www.croiconference.org/sessions/antiviral-activity-efda-against-nrti-sensitive-and-resistant-strains-hiv-2 - Grobbler J et al MK-8591 concentrations at sites of HIV transmission and replication. CROI 2017, 13-16 February, Seattle, Washington. Poster abstract 435.
http://www.croiconference.org/sessions/mk-8591-concentrations-sites-hiv-transmission-and-replication - Matthews RP. Single doses as low as 0.5 mg of the novel NRTTI MK-8591 suppress HIV for at least seven days. IAS 2017, Paris. Late breaker poster abstract TUPDB020.
http://programme.ias2017.org/Abstract/Abstract/5525 - Markowitz M et al. Weekly oral MK-8591 protects male rhesus macaques against repeated low dose intrarectal challenge with SHIVC109P3. IAS 2017, Paris. Late breaker oral abstract MOAX0203LB.
http://programme.ias2017.org/Abstract/Abstract/5533 - Cihlar T et al. GS9148: A novel nucleotide active against HIV-1 variants with drug-resistance mutations in reverse transcriptase. 13th CROI, 5-8 February 2006, Denver. Oral abstract 45. (Abstract no longer online).
- Cihlar T et al. Design and profiling of GS-9148, a novel nucleotide analog active against nucleoside-resistant variants of HIV Type 1, and its orally bioavailable phosphonoamidate prodrug, GS-9131. Antimicrob Agents Chemother. 2008 Feb; 52(2): 655–665. Published online 2007 Dec 3. doi: 10.1128/AAC.01215-07.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2224772 - White KL et al. GS-9131 is a novel NRTI with activity against NRTI-resistant HIV-1. CROI 2017, 13-16 February, Seattle. Poster abstract 436.
http://www.croiconference.org/sessions/gs-9131-novel-nrti-activity-against-nrti-resistant-hiv-1 - ViiV Healthcare press statement. Medicines in development. (December 2015).
https://www.viivhealthcare.com/our-medicines/medicines-in-development.aspx - Collins S. GSK discontinues development of maturation inhibitor BMS-955176. HTB, (October 2016).
https://i-base.info/htb/30865 - Joshi SR et al. An analysis of neurologic and psychiatric adverse events of subjects receiving the investigational HIV-1 maturation inhibitor (MI) GSK3532795/BMS-955176. IAS 2017, Paris. Poster abstract WEPEB0540.
http://programme.ias2017.org/Abstract/Abstract/2917 - Krystal M et al. HIV combinectin GSK3732394: a long-acting inhibitor with multiple modes of action. HIV Glasgow 2016. Poster abstract P022.
http://www.natap.org/2016/GLASGOW/GLASGOW_27.htm - Link JO et al. Novel HIV PI with high resistance barrier and potential for unboosted QD oral dosing. CROI 2017, 13-16 February, Seattle. Late breaker abstract 433.
http://www.croiwebcasts.org/p/2017croi/croi33636 - Tse WC et al. Discovery of novel potent HIV capsid inhibitors with long-acting potential. CROI 2017, 13-16 February, Seattle, Washington. Oral abstract 38.
http://www.croiconference.org/sessions/discovery-novel-potent-hiv-capsid-inhibitors-long-acting-potential - Wu H et al. Efficacy and safety of long-acting HIV fusion inhibitor albuvirtide in antiretroviral- experienced adults with HIV-1: interim 48-week results from the randomised, controlled, phase 3, non-inferiority TALENT study. HIV Glasgow 2016, 23-26 October. Oral abstract O336.
https://vimeo.com/189136480 (webcast) - Murphy R et al. Elsulfavirine as compared to efavirenz in combination with TDF/FTC: 48-week study. CROI 2017, 13-16 February, Seattle. Late breaker abstract 452LB.
http://www.croiconference.org/sessions/elsulfavirine-compared-efavirenz-combination-tdfftc-48-week-study - Bichko V et al. Pre-clinical pharmacokinetics of elsulfavirine/VM1500A long acting injectable formulations. IAS 2017, 23-26 July, Paris. Poster abstract WEPEA0190.
http://programme.ias2017.org/Abstract/Abstract/1515 - Mitchell ML et al. Novel non-catalytic site integrase inhibitor with improved resistance profile. CROI 2017, 13-16 February, Seattle. Poster abstract 434.
http://www.croiconference.org/sessions/novel-non-catalytic-site-integrase-inhibitor-improved-resistance-profile - Scherrer D et al. Early evidence of antiviral activity & safety of ABX464 in HIV treatment-naïve patients. CROI 2016, February 22–25, Boston, Late breaker poster abstract 461LB.
http://www.croiconference.org/sites/default/files/posters-2016/461LB.pdf (PDF) - Differential efficacy of ABX464 and its primary metabolite ABX464-NGlc on HIV replication: implications for treatment strategies to eliminate viral reservoirs. HIV Glasgow, 23–26 October, 2016.
http://www.natap.org/2016/HIV/060116_02.htm - Paredes R et al. ABX464 decreases total HIV DNA in PBMC´s when administered during 28 days to HIV-infected patients who are virologically suppressed. IAS 2017, 23-26 July, Paris. Poster abstract TULBPEB22.
http://programme.ias2017.org/Abstract/Abstract/5650